

I had the opportunity to be a contributor to Google Summer of Code 2022, working with DeepChem (Open Chemistry), a project that aims to create high-quality, open-source tools to democratize the use of deep learning in drug discovery, materials science, quantum chemistry, and biology.
Among its suite of various machine learning models, it has a wide range of graphs-based neural network model implementations aimed to solve the applications such as predicting the solubility of small drug-like molecules, binding affinity for a small molecule to protein targets, analyzing protein structures, and extracting useful descriptors.
Through this forum post, I would like to comprehensively document and describe all of my work done over the summer.
Project Title
D-MPNN Model Implementation for DeepChem
This project aimed to implement a Directed — Message Passing Neural Network (D-MPNN) [1] model, a graph convolution network (GCN) built upon the existing Message Passing Neural Network (MPNN) model based on the base implementation in Chemprop.
Some Important Links
My GitHub: https://github.com/ARY2260
DeepChem Home Page: https://deepchem.io/
DeepChem GitHub: https://github.com/deepchem/deepchem
Proposal Link: Link
Preface
Molecular property prediction, one of the oldest cheminformatics tasks, has received new attention in light of recent advancements in deep neural networks. These architectures either operate over fixed molecular fingerprints common in traditional QSAR models, or they learn their own task-specific representations using graph convolutions. Both approaches are reported to yield substantial performance gains, raising state-of-the-art accuracy in property prediction.
The Analyzing Learned Molecular Representations for Property Prediction [1] paper introduced the D-MPNN algorithm for property prediction that outperforms these existing strong baselines across a range of data sets. The model has two distinctive features:
- It operates over a hybrid representation that combines convolutions and descriptors. This design allows it to learn a task-specific encoding while providing a strong prior with fixed descriptors.
- It learns to construct molecular encodings by using convolutions centered on bonds instead of atoms, thereby avoiding unnecessary loops during the message passing phase of the algorithm.
Contents covered in the forum
– Expected Deliverables
– Featurization of molecule SMILES
– Directed-MPNN model
– Model Benchmark Results
– Usage
– Fixes and Additions to DeepChem
– Future Scope
– Acknowledgement
– References
Expected Deliverables
Implementation of classes and support functions for featurization of SMILES data.
Implementation of additional loss classes. (not required for DeepChem)
Model training and benchmarking. (In Progress)
Featurization of molecule SMILES
Cheminformatics datasets contain molecules represented as SMILES. These SMILES can be analysed using the RDKit library to get information about the atoms and bonds in the molecules.
Chemical structures and their respective SMILES
Molecular fingerprinting is a vectorized representation of molecules capturing precise details of atomic configurations. During the featurization process, a molecule is decomposed into substructures (e.g., fragments) of a fixed-length binary fingerprint assembled into an array whose each element is either 1 or 0.
For this project, I implemented atomic and bond-level featurization and molecule-level (global) featurization in DeepChem, specific to D-MPNN model requirements.
Atomic and Bond-level featurization
The D-MPNN paper [1] suggested 133 features for each atom and 14 features for each bond in a molecule.
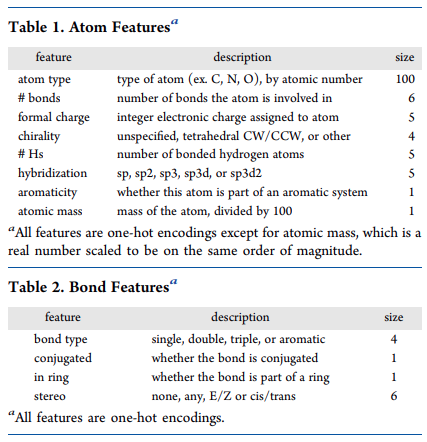
The individual features are extracted from SMILES using RDKit library and one-hot encoded to get vectorized representation.

Corresponding Pull Requests:-
- atom features function and helper functions for DMPNN Featurizer #2929 (Merged) +257 −0
- add bond_features and reaction mapping with suitable tests for D-MPNN #2942 (Merged) +221 −6
- modify molecular featurizer base class and suitable tests #2960 (Merged) +150 −6
- added _MapperDMPNN class and suitable tests #2962 (Merged) +280 −14
Molecule-level (global) featurization
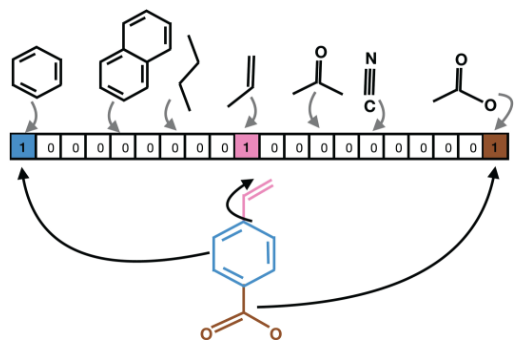
The molecule-level fingerprints describe the properties, like molecular weight, number of valence electrons, maximum and minimum partial charge, etc. Options available for global feature generators:
- 2048 — Morgan Fingerprints (and count-based)
- 200 — RDKit descriptors (normalized)
I modified the morgan fingerprint generator to include an option to generate count-based fingerprints. I also modified RDKitDescriptors() class in DeepChem to compute normalized descriptors. This functionality is based on the implementation of the RDKit2DNormalized() method in Descriptastorus library.
The neural network architecture requires that the features are appropriately scaled to prevent features with large ranges from dominating smaller ranged features, as well as preventing issues where features in the training set are not drawn from the same sample distribution as features in the testing set. To prevent these issues, a large sample of molecules is used to fit cumulative density functions (CDFs) for all features. CDFs were used as opposed to simpler scaling algorithms mainly because CDFs have the useful property that each value has the same meaning: the percentage of the population observed below the raw feature value.
Corresponding Pull Requests:-
- add global feature generator and suitable unit tests #2971 (Merged) +149 −0
- add count-based morgan fingerprint featurizer and suitable unit tests #2980 (Merged) +61 −30
- modify RDKitDescriptors class for normalized features #2983 (Merged) +894 −59
- add new global feature generators and units tests for DMPNN featurizer #3005 (Merged) +74 −23
Pull request for main featurizer class for the D-MPNN model: (Merged)
The DMPNNFeaturizer() class is the main featurizer class for the D-MPNN model. The _featurize() method has been defined here, which takes a datapoint (RDKit molecule) as input and returns a GraphData() class instance for that datapoint.
Directed-MPNN model
Directed — Message Passing Neural Network (D-MPNN) model is a graph convolution network (GCN) built upon the existing Message Passing Neural Network (MPNN) architecture. The primary difference between the D-MPNN and regular MPNNs is in the nature of the messages being passed through the molecule during the message passing phase. While the general MPNN framework assumes messages are centered on atoms , the D-MPNN centers messages on bonds instead.
Specifically, the D-MPNN maintains two representations for the message centered on the bond between atoms 𝑣 and 𝑤 : one from atom 𝑣 to atom 𝑤 and one from atom 𝑤 to atom 𝑣 , hence the word Directed . Consequently, rather than aggregating information from neighboring atoms, the D-MPNN aggregates information from neighboring bonds. Each bond’s message is updated based on all incoming bond messages.
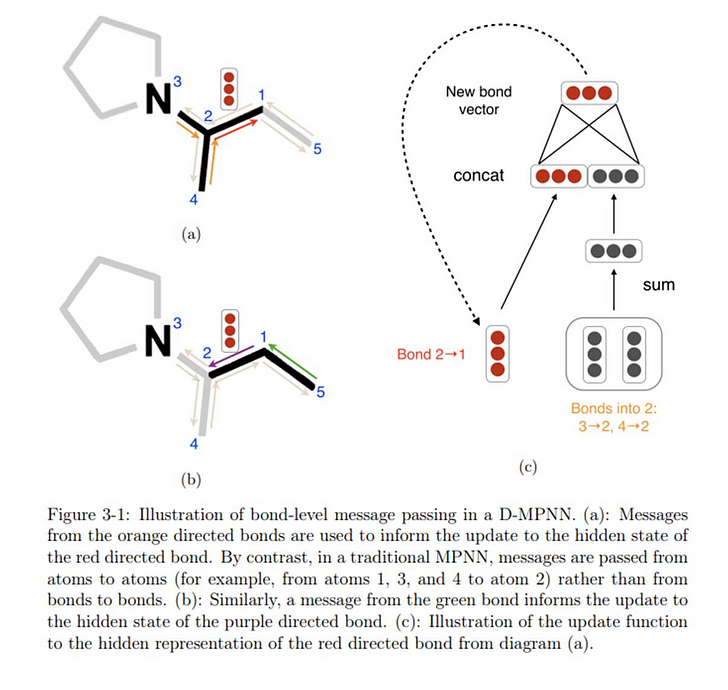
The motivation of this design is to prevent totters, that is, to avoid messages being passed along any path of the form v1 v2 ··· vn where vi = vi+2 for some i. Such excursions are likely to introduce noise into the graph representation. Due to this structure, with messages centered on bonds and a distinction between the two directions of bond messages, the D-MPNN has greater control over the flow of information across the molecule and can therefore build more informative molecular representations.
Implementation
The D-MPNN model has 2 phases, the message-passing phase and the read-out phase.
- The message-passing phase aims to generate ‘hidden states of all the atoms in the molecule’ using encoders.
- Next, the features are passed into a feed-forward neural network , in the read-out phase to get the task-based prediction.
For the message-passing phase, I initially created the _MapperDMPNN() class which returns modified features based on the given GraphData() object generated from the featurizer for each molecule.
add mapper class for dmpnn model and suitable unit tests #3001 (Merged) +313 −0
Fix issue #3057 (update _Mapper class for dmpnn) #3058 (issue: #3057) (Merged)
Then, I implemented the DMPNNEncoderLayer() class, a derivative torch.nn.Module class. It takes a batch of modified features from the _MapperDMPNN() class to generate convoluted molecular encodings concatenated with global features (if any).
add dmpnn encoder layer and suitable unit test #3023 (Merged) +414 −1
Next, for the feed-forward neural network (ffn) used in the readout phase, I modified the PositionwiseFeedForward() class in DeepChem to contain a dropout_at_input_no_act condition to initialize the ffn with a dropout layer at the input when used for D-MPNN model.
modify PositionwiseFeedForward class and add unit tests #3009 (Merged) +33 −2
The D-MPNN algorithm is implemented in D-MPNN() class which defines the various encoder layers and establishes a sequential model. The number of encoders required is equal to the number of SMILES columns in the dataset. The class supports two modes: regression and classification .
add dmpnn class and suitable unit tests #3028 (Merged) +387 −1
Finally, I implemented the DMPNNModel() class (derivative of TorchModel() class), which is a wrapper class for DMPNN(). It is the primary model class that the user will use to initialize the D-MPNN model that handles training, interpretations, saving, and reload operations.
add torch model wrapper for DMPNN model class #3034 (Merged) +341 −2
The batching functionality was implemented later on using the PyTorch-Geometric library. It was a relatively challenging task for me to perform encoding over a batch of graph data from the molecules. The solution was to create a concatenated tensor (with suitable increment operations) for each feature in a batch of molecules. Later, when the D-MPNN encoder generates a single tensor containing encodings for that batch, it is split into respective tensors for individual molecules using a list of sizes of all the molecules in that batch.
implementation of batching for DMPNN model #3040 (Open) +290 −129
Model Benchmark Results
In the last phase of this project, I benchmarked the implemented model with two datasets, Delaney (regression) and Tox21 (classification). I compared this model against the Chemprop implementation and graph convolution in DeepChem. The results are averaged over three runs.
Chemprop suggested the following hyperparameter configuration used for benchmarking:
{
“depth”: 6,
“enc_dropout_p”: 0.0,
“enc_hidden”: 1800,
“ffn_dropout_p”: 0.0,
“ffn_hidden”: 1800,
“ffn_layers”: 2,
“learning_rate”: 1e-4
}
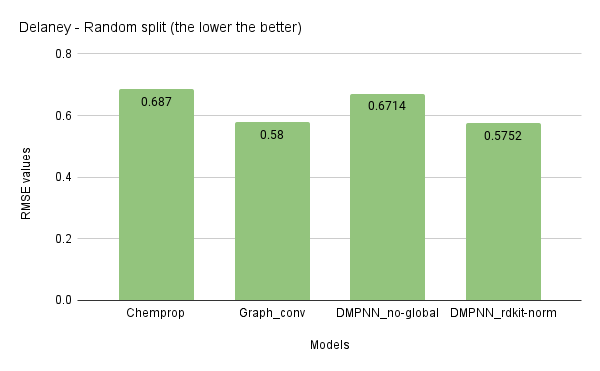

The above benchmarks show that the D-MPNN model results are similar to that of the Chemprop. The benchmarking of other datasets is still in progress while writing this forum post. Pending datasets include: ESOL ®, FreeSolv ®, QM8 ®, QM9 ®, SIDER ©, MUV ©, HIV ©, BBBP ©, PCBA ©. (R — Regression, C — Classification)
I have also tested the D-MPNN model with scaffold split:
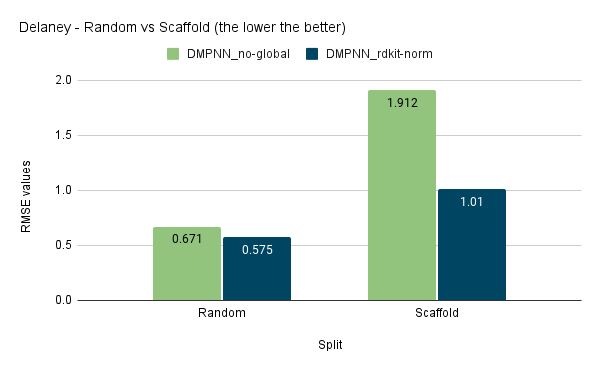
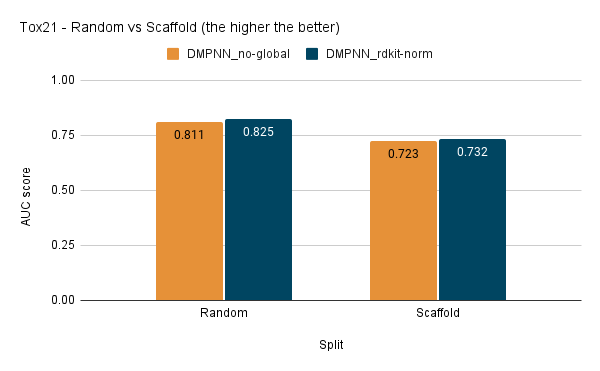
It is interesting to note that the use of global featurizer like RDKit Normalised Descriptors has provided better regression results and made the model more generalized. Still, the difference is not much significant in the case of classification results.
Inferring from the results so far, the implementation seems robust, and I look forward to other benchmarks.
Usage
The script below is an example of how to use this model:
import deepchem as dc
from deepchem.models import DMPNNModel
# Load Tox21 dataset
tox21_tasks, tox21_datasets, transformers = dc.molnet.load_tox21(featurizer=dc.feat.DMPNNFeaturizer(features_generators=["rdkit_desc_normalized"]), splitter='scaffold')
train_dataset, valid_dataset, test_dataset = tox21_datasets
print('dataset is featurized')
# Assess featurized data
print(len(train_dataset), len(valid_dataset), len(test_dataset))
print(train_dataset.X[:5])
# Initialise the model
model = DMPNNModel(n_tasks=len(tox21_tasks), \
n_classes=2, \
mode='classification', \
batch_size=50, \
global_features_size=200)
# Model training
print("Training model")
model.fit(train_dataset, nb_epoch=30)
# Model evaluation
metric = dc.metrics.Metric(dc.metrics.roc_auc_score, np.mean)
print("Evaluating model")
train_scores = model.evaluate(train_dataset, [metric], transformers)
valid_scores = model.evaluate(valid_dataset, [metric], transformers)
print("Train scores: ", train_scores)
print("Validation scores: ", valid_scores)
Fixes and Additions to DeepChem
I have made a total of 18 PRs, 40+ commits, and 4500+ additions during this summer .
DeepChem suite now includes:
- A fully functional SMILES featurizer for the D-MPNN model with support for global molecular features.
- Count-based Morgan fingerprint featurizer.
- An upgraded RDKit descriptors generator, now with the ability to generate normalized descriptors.
- A new Encoder layer class and a modified Feed Forward class to support D-MPNN functionality.
- A new torch model class for D-MPNN, with support for batching.
Pull Requests to solve issues:
- resolved deprecation warning #2937 (issue: #2936) (Merged)
- Fixed a VisibleDeprecationWarning is reported for running doctest on graph_feature.py file.
- fix bug in GraphData class and add suitable unit test #2979 (issue: #2978) (Merged)
-
GraphData()class checks if theedge_indexcontains the invalid node number, using the conditionnp.max(edge_index) >= len(node_features). In case of single atom molecules,edge_indexin an empty array ofshape = (2, 0)and np.max() method raises an error for empty array :Error: zero-size array to reduction operation maximum which has no identity. -
np.max()method works only for a non-empty array, so the array size should be non-zero. This condition is added in theGraphDataclass through this PR.
Next, I plan to add a tutorial to use the D-MPNN model.
You can find my progress reports, made throughout the summer, for the project at the DeepChem forum.
Future Scope
A potential upgrade to the D-MPNN model could be the implementation of a directed graph-based Communicative Message Passing Neural Network (CMPNN) based on [2] to improve the molecular graph embedding by strengthening the message interactions between bonds and atoms. Its code would be a derivative of the current implementation of the D-MPNN model.
Acknowledgment
I express immense gratitude for all the support extended by my mentors Bharath Ramsundar , Arun , Tony Davis , Stanley Bishop , and the DeepChem community for their suggestions and discussions.
I am happy to have found an organization where I could learn a lot about the real-world workings of ML models in sophisticated expert systems, primarily using graph-based neural networks. I plan to continue contributing to DeepChem by improving the project implementation while also fixing issues and helping the community wherever and whenever possible!
Thank you, Google, for this fantastic opportunity.
References
[1] Analyzing Learned Molecular Representations for Property Prediction
[2] Communicative representation learning on attributed molecular graph
You have reached the end of this forum post. Thank you for your valuable time.