
I recently read the paper Unified rational protein engineering with sequence only deep representation learning. The authors have released their source code at https://github.com/churchlab/UniRep.
The authors’ driving motivation is to improve the state of the art in protein engineering. A lot of traditional protein engineering is reliant on random variation without systematic modeling of the relationship between sequence and function. Or alternatively, on expensive structure based modeling techniques that use molecular dynamics or free energy calculations. As deep learning techniques mature, the dream is that we might be moving closer to practical and cheap rational guided protein design. This work takes a nice step in that direction.
The core contribution of the paper is a methodology to turn a protein sequence into an embedded feature vector in an unsupervised fashion. While past papers have considered alternative deep architectures for protein engineering, these methods have mostly been supervised, which means the applicability of the techniques is limited by the scarcity of labeled data. The authors show that their unsupervised embedding vectors can be used for a number of downstream tasks, including the prediction of the stability of natural and de novo designed proteins. Here’s a figure that walks through the core methodology of the paper.
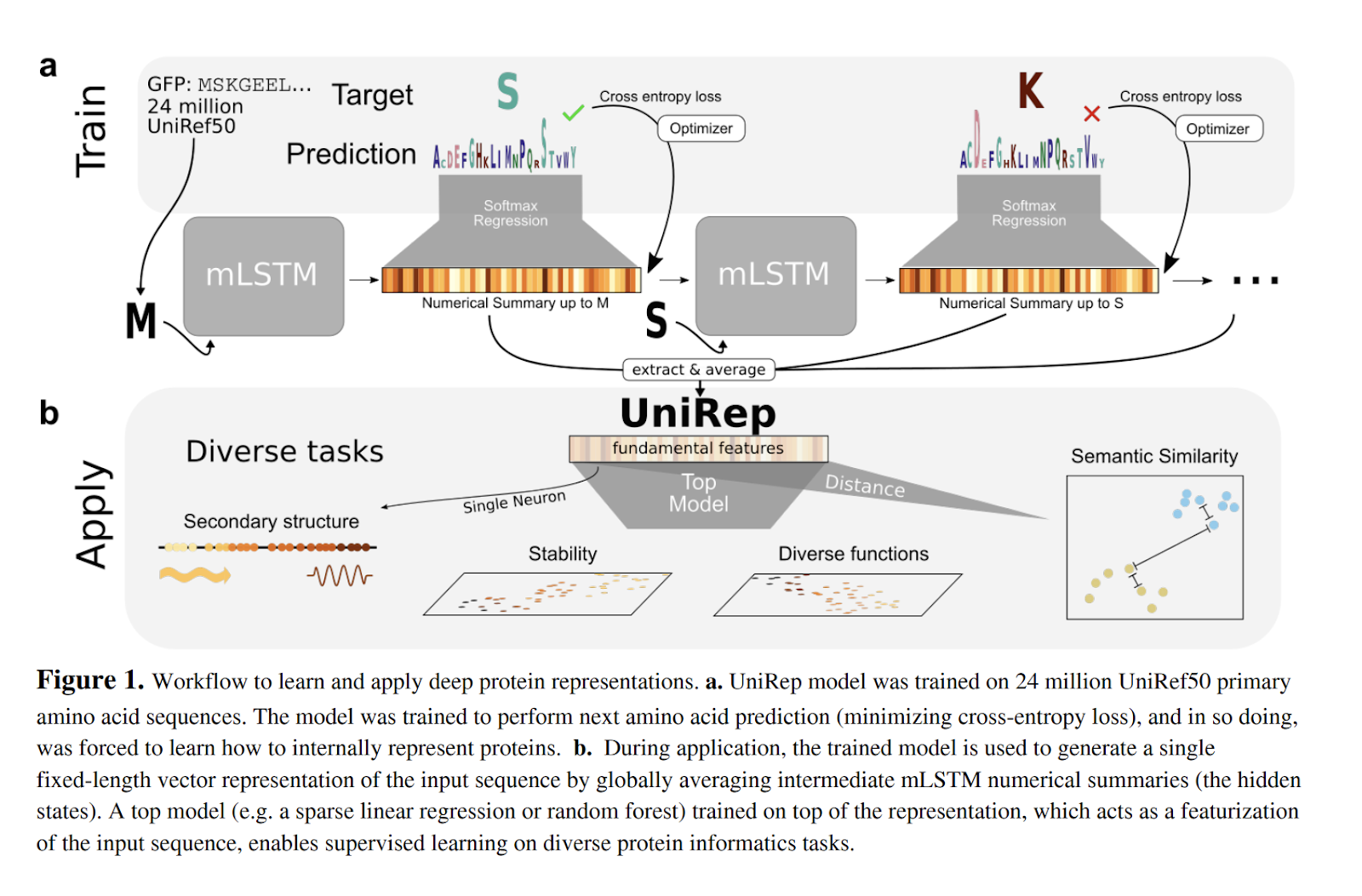
The models themselves are multiplicate LSTMs, trained with data from Uniref. The models are trained with an unsupervised loss, on the task of predicting next characters in the sequences on a dataset of 24 million Uniref50 sequences.
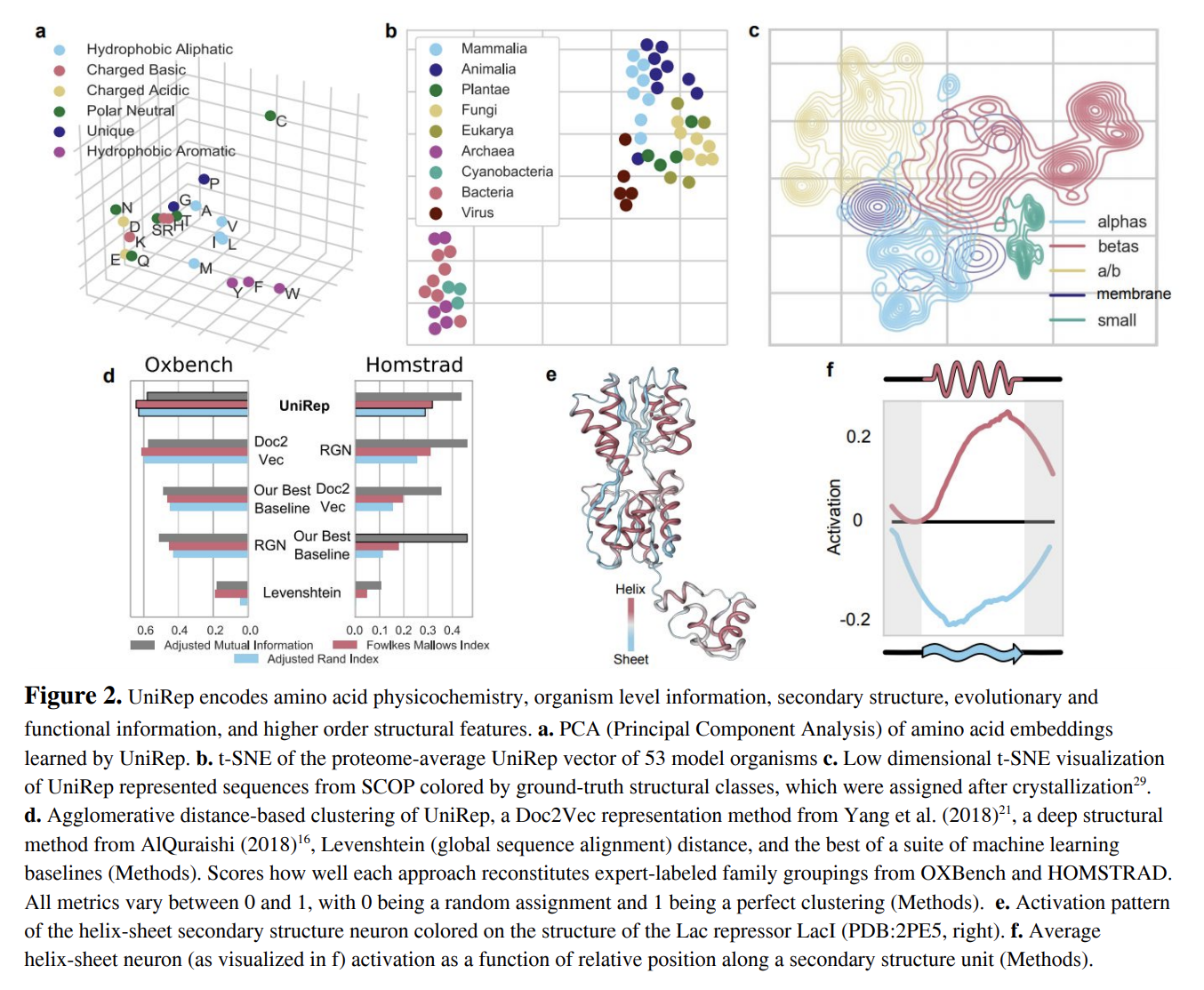
The paper does a wide range of analysis of the learned representations. A t-SNE analysis on on proteome clusters finds meaningful clusters in proteome data depending on organism type (Figure 2b). Interestingly, figure 2c above demonstrates that the model is able to structurally separate proteins from the Structural Classification of Proteins (SCOP) derived from crystallographic data. In a twist reminiscent of OpenAI’s sentiment neuron, the authors discover a single neuron in their trained model that discriminated beta sheets from alpha helices (Figures 2e, 2f). This tidbit is really interesting to me since it provides a compelling demonstration that geometric properties can be learned from the raw sequences in unsupervised fashion.
The authors continue by analyzing stability measurements for de novo designed mini proteins. The paper benchmarks against other ML algorithms and against Rosetta (Figure 3a-3d below). Simple linear models are trained on top of Unirep vectors and compared against other methods from the literature. Strong results are achieved on a suite of benchmark tasks, which leads the authors to argues that results provide justification that UniRep approximates fundamental biophysical features shared between all proteins
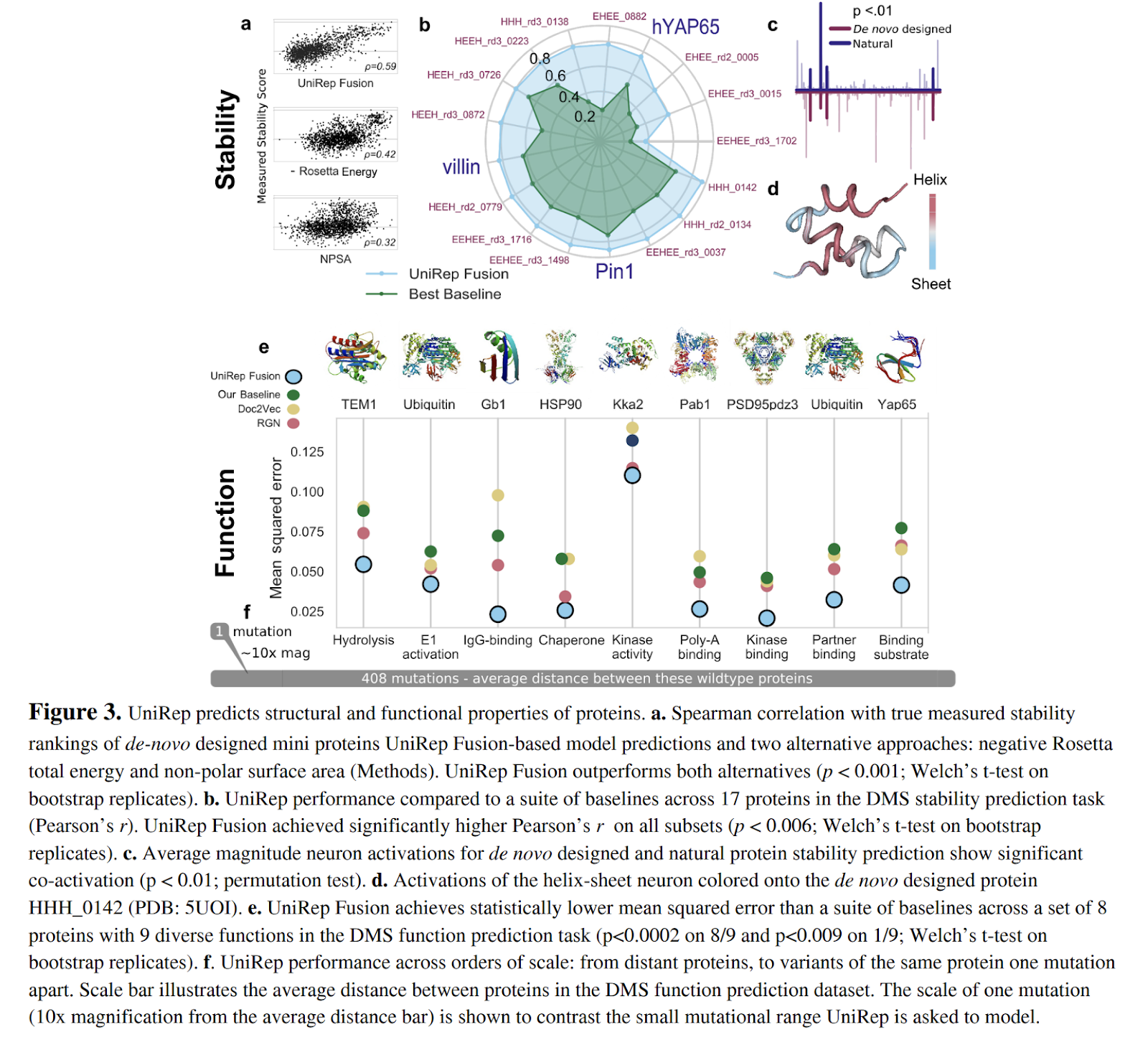
As a further test of the Unirep representation vectors, the authors try applying the vectors to the task of predicting protein function directly from sequence (Figure 3e-3f above). They source a collection of quantitative function prediction datasets which feature single point mutants of wildtype proteins (recall that a single point mutant takes the naturally occurring “wildtype” protein and applies a mutation to a single amino acid in the protein). Simple linear models on Unirep offer improvements over baseline methods.
The authors investigate the capability of Unirep models to generalize broadly. Since Unirep is trained on a wide assortment of protein data, the hope is that the models might generalize powerfully. The tradeoff here is that the general applicability might mean a lack of local focus. The paper proposes the use of a technique they call evotuning (evolutionary fine tuning) to try to get the best of both worlds. They used JackHMMER search to find a set of 25K sequences that are likely evolutionarily linked. The general Unirep model is then trained with an unsupervised next-character prediction task on this evolutionarily linked dataset .
These representations are then tested on their ability to predict the phenotype (brightness) of distant Aequorea victoria green fluorescent protein (avGFP) variants given limited information about local variants.
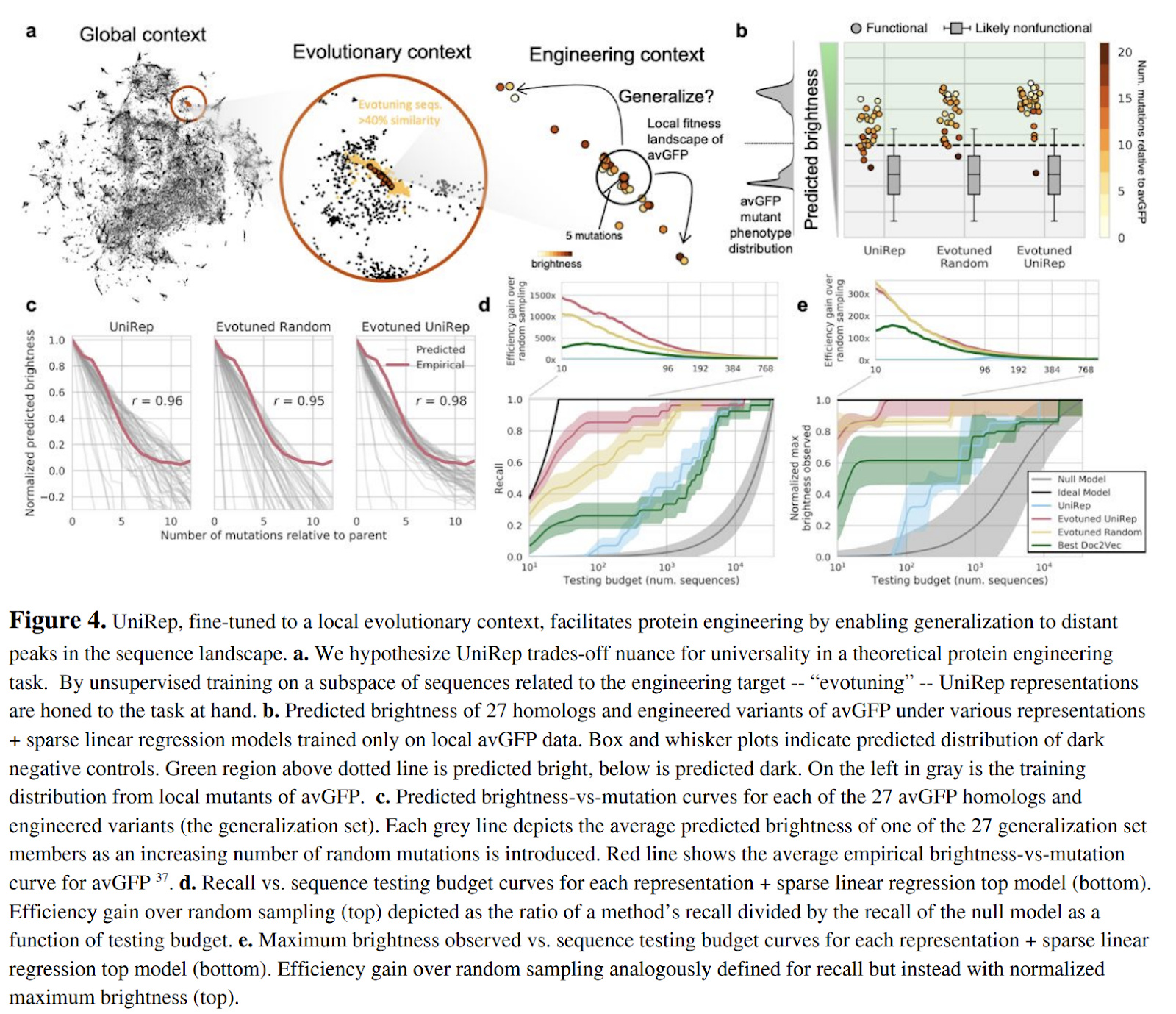
Figure 4c shows some performance improvements of Evotuned UniRep over the baseline UniRep and Evotuned Random (evotuning starting from random weights), but the improvements appear to be relatively marginal.
The evotuned UniRep model was then put to work on the actual task of discovering bright GFP homologs. Figures 4d and 4e above show strong performance over the null model, with some improvements on stronger baselines as well. Interestingly, improvements are particularly robust in the small data regime.
The appendix has some additional details on the training done to build models. The authors did some manual exploration with other classes of RNNs past the mLSTM but settled on mLSTM after experimentation. This paper is from a few years ago, which makes me think that repeating these experiments with the latest BERT based transformer models would likely yield stronger results. I’d bet that someone has already done this, or is in the process of doing this right now.
The authors mention an intriguing throwaway early in the paper that coevolutionary methods behave poorly in underexplored parts of protein space. This caught my attention since AlphaFold makes heavy use of co-evolutionary information. I wonder if UniRep style representations could be used to juice AlphaFold style techniques to be more powerful. The authors hint at this in their discussion, by suggesting that UniRep could be combined with recurrent geometric networks to advance ab-initio protein structure prediction tasks. I wonder if they’re working on this idea at present.
All in all, I quite liked this paper. The strong results established here make me think that unsupervised learning could have some very intriguing impacts on bioinformatics and protein/antibody design in the years to come.