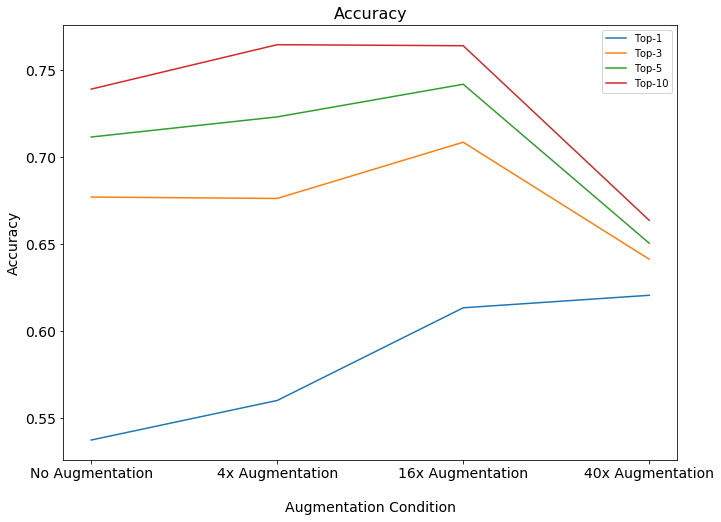
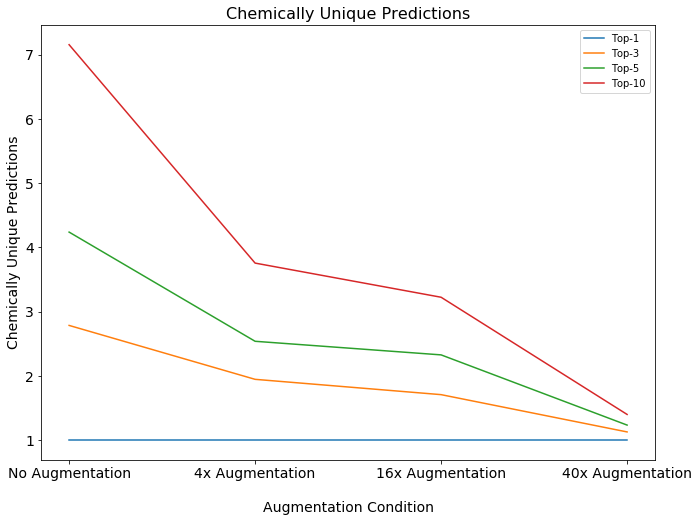
There have been some considerable advances in data augmentation. This recent paper from Google research shows very impressive results through clever use of unsupervised data augmentation which combines augmentation with a new loss function that encourages consistency of labeling for transformed versions of the original sample.
How could we adapt these techniques to molecular data? It feels like it ought be able able to create data augmented versions of molecules and use the unsupervised data augmentation loss to allow for higher accuracies on small molecular datasets.