
I recently read the article Ultra-large library docking for discovering new chemotypes. This is a paper that I’ve been meaning to read for some time. The use of molecular docking at scale to guide the discovery of novel therapeutic compounds is something that’s become increasingly common as the ease of working with cloud systems grows. This current paper is from a group that’s been working with docking for quite a while at UCSF.
Before we dive into the papers, here’s a quick note about source code. The source for the full paper wasn’t released (perhaps because some of the analysis was done human-in-the-loop), but there are a few open resources the authors have made available:
- https://github.com/docking-org: The top level GitHub repo for the authors.
- https://github.com/docking-org/ChemInfTools - Some chemoinformatic tools used in the paper
- http://star.mit.edu/cluster/ - The paper seems to use the star cluster framework to simplify launching jobs on AWS
- https://github.com/rasbt/smilite - Tools to look up compounds from Zinc. Useful for pulling down compounds the authors reference.
The paper performs structured based docking of 170 million make on demand compounds from 130 well characterized reactions. Molecules are docked against AmpC \beta-lactamase and D_4 dopamine receptor. For each receptor, 44 and 549 compounds were synthesized and tested. A phenolate inhibitor of AmpC (a group of inhibitors without precedent) was discovered in the course of this screen. If you haven’t refreshed your organic chemistry in a bit, here’s a quick reference picture of a phenolate:
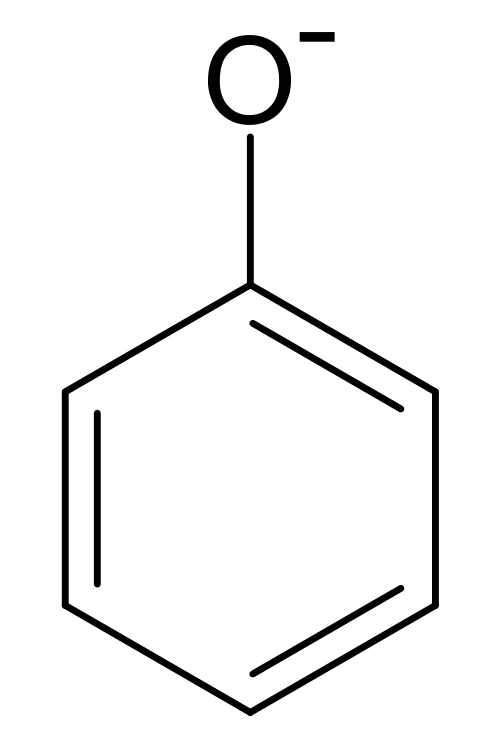
The lead compound was optimized to 77nm, making it one of the most potent known non-covalent binders. Co-crystal structures of this compound (and others) confirmed docking predictions
As a brief bit of more general commentary, this work takes a different tack from other recent computational drug discovery papers, such as this recent paper from Google. That work limits the chemical space tested to a particular DNA encoded library, using the fact this library can be densely sampled to effectively train a deep learning system that’s predictive. This work on the other hand doesn’t limit itself to a given chemical library, but rather samples broadly using the simpler physics-inspired docking scoring function to guide its search.
The tradeoff here is that docking scoring is known to be inaccurate though, so many false positives. Chemical space is vast, so the work focused on molecules from 130 well characterized reactions using 70K building blocks from enamine. Here’s a figure that looks at some of the example building blocks used and the example products.
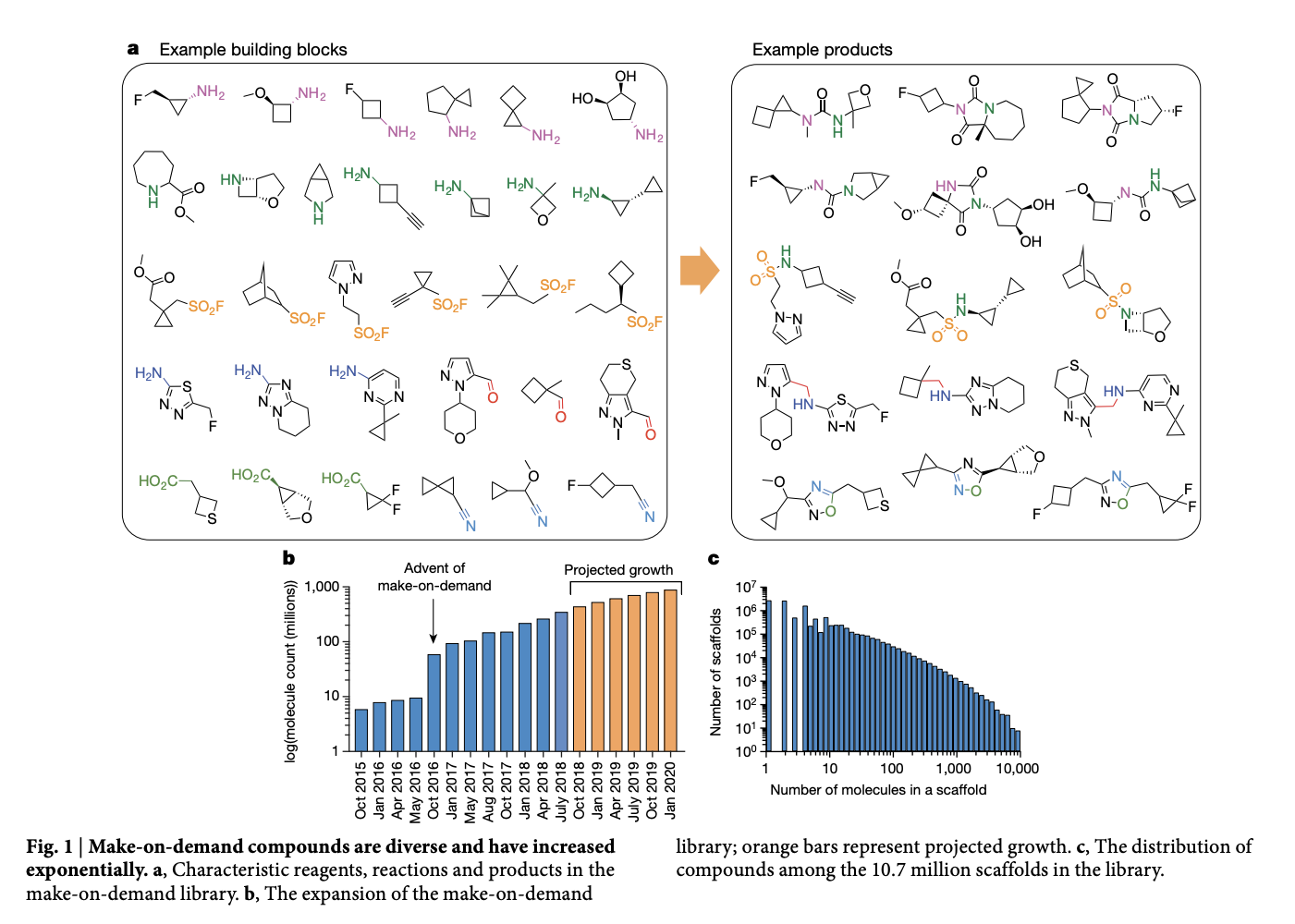
Each compound was fit into the enzyme active site with an average of 4,054 orientations. For each orientation, 280 conformations were sampled. The complex was rigid body minimized with a simplex minimizer. As a quick bit of commentary, that’s a lot of orientation-conformation pairs for each compound. I suspect that this heavy sampling was critical though in making the molecular docking yield reasonable results.
70 trillion complexes were sampled in the orthosteric site of the \textrm{D}_4 receptor, for a total of 44K CPU hours, run in 1.2 physical days on 1500 cores. I really wish the authors had open sourced the source code for this final calculation. This calculation seems like it would have required some good cloud infrastructure, and best practice example code would be useful to have.
Molecules similar to known dopaminergic, serotonergic, adrenergic ligands were removed as part of the cleanup step. The authors used visual inspection to remove some additional “bad” compounds. 44 and 549 compounds were synthesized for AmpC and \textrm{D}_4 Dopamine respectively. Crystallography was used to find the true binding poses of the some of the predicted inhibitors as the image below shows:
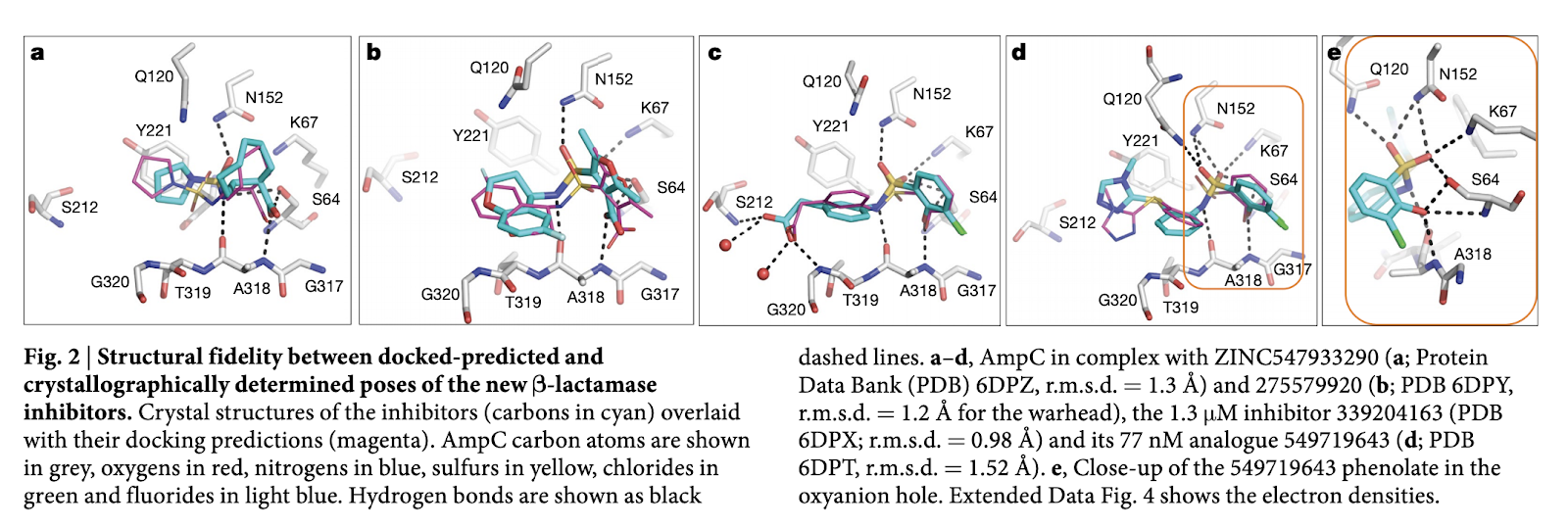
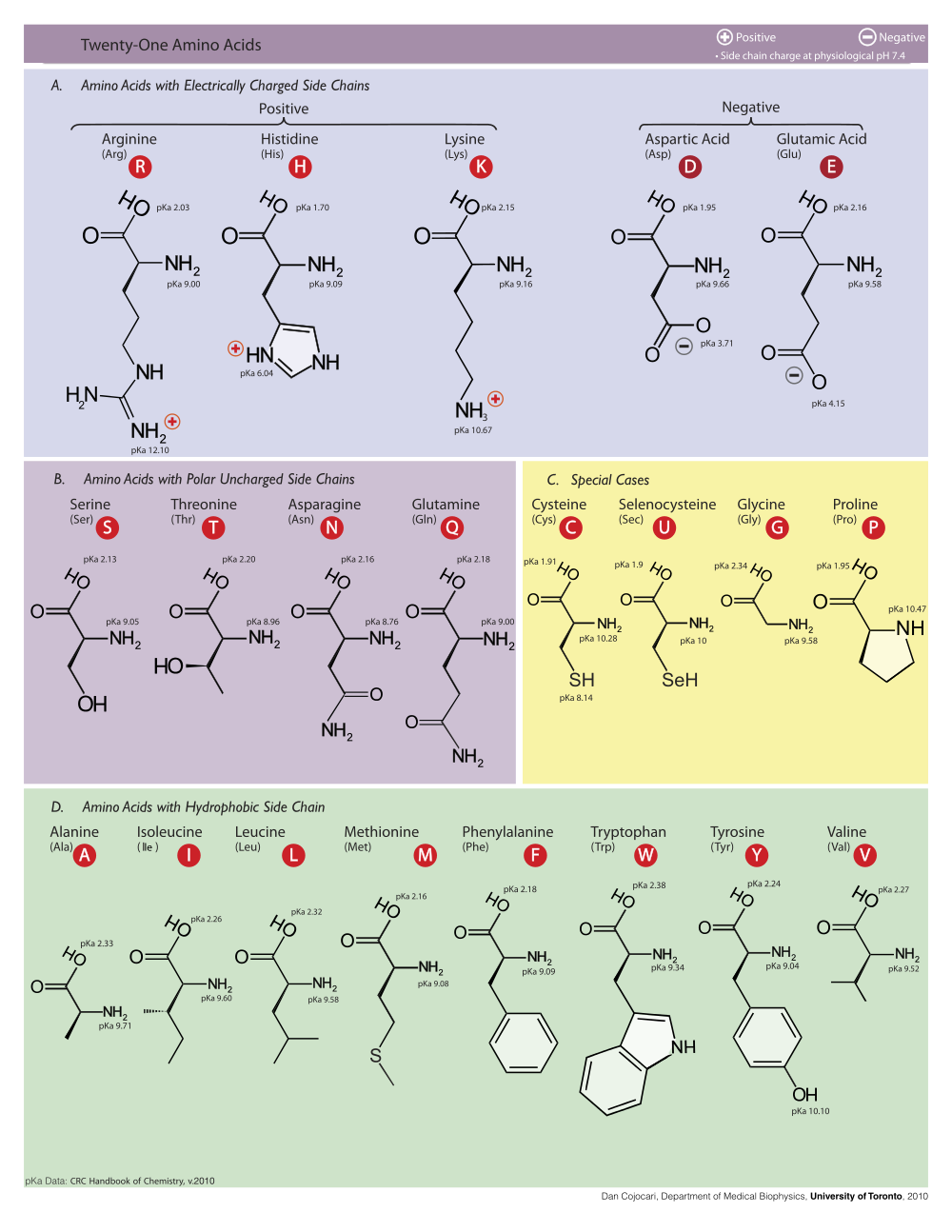
Reading images of this type can be tough. The major thing you need to have is a working knowledge of amino acids. If you haven’t looked at these types of diagrams before, I’d recommend matching the amino acids against those in the chart below and trying to build some intuition about the rough geometry of the binding pocket.
This next diagram tells an interesting story. Docking score does appear somewhat correlated with activity, but it’s clearly a relatively loose prediction. The paper demonstrates that it can work quite well nonetheless!
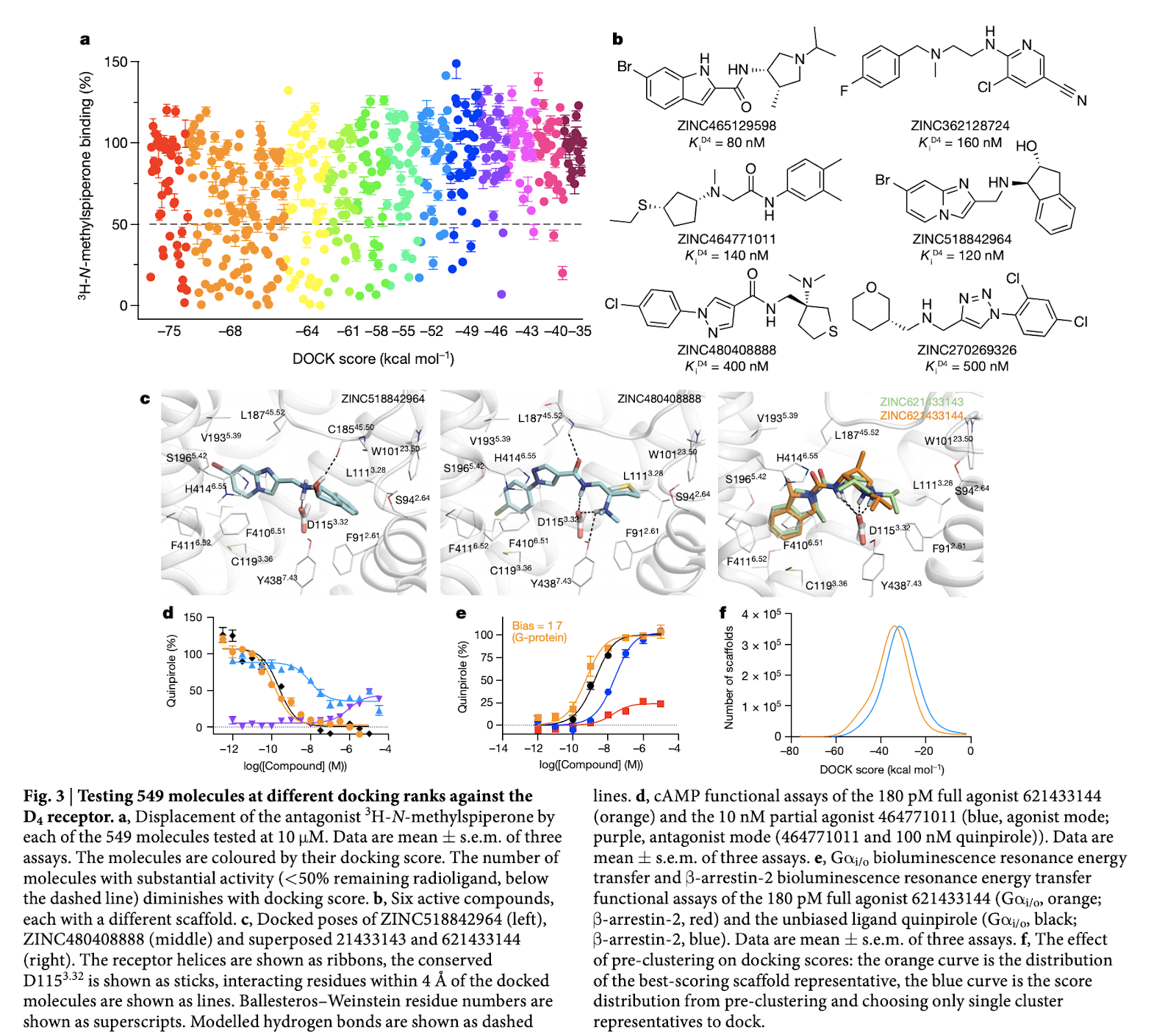
The total process in the work results in hit rates around 24%. It’s interesting to compare this number with the 30% hit rate from the recent Google paper. There are different strengths/weaknesses to both techniques. This paper’s docking based approach requires knowledge of the target structure up front, and knowledge of the active site of the protein, but no experimental assay data is needed to start. For Google’s technique, the protein target doesn’t have to be known up front, but experimental data is required to bootstrap the model instead.
The authors do some interesting additional analysis to try to determine the total number of active compounds in the full library. This is a useful bit of analysis that might be of interest to a team deciding whether they should go to the effort of doing a high throughput assay. The figure below has some more information
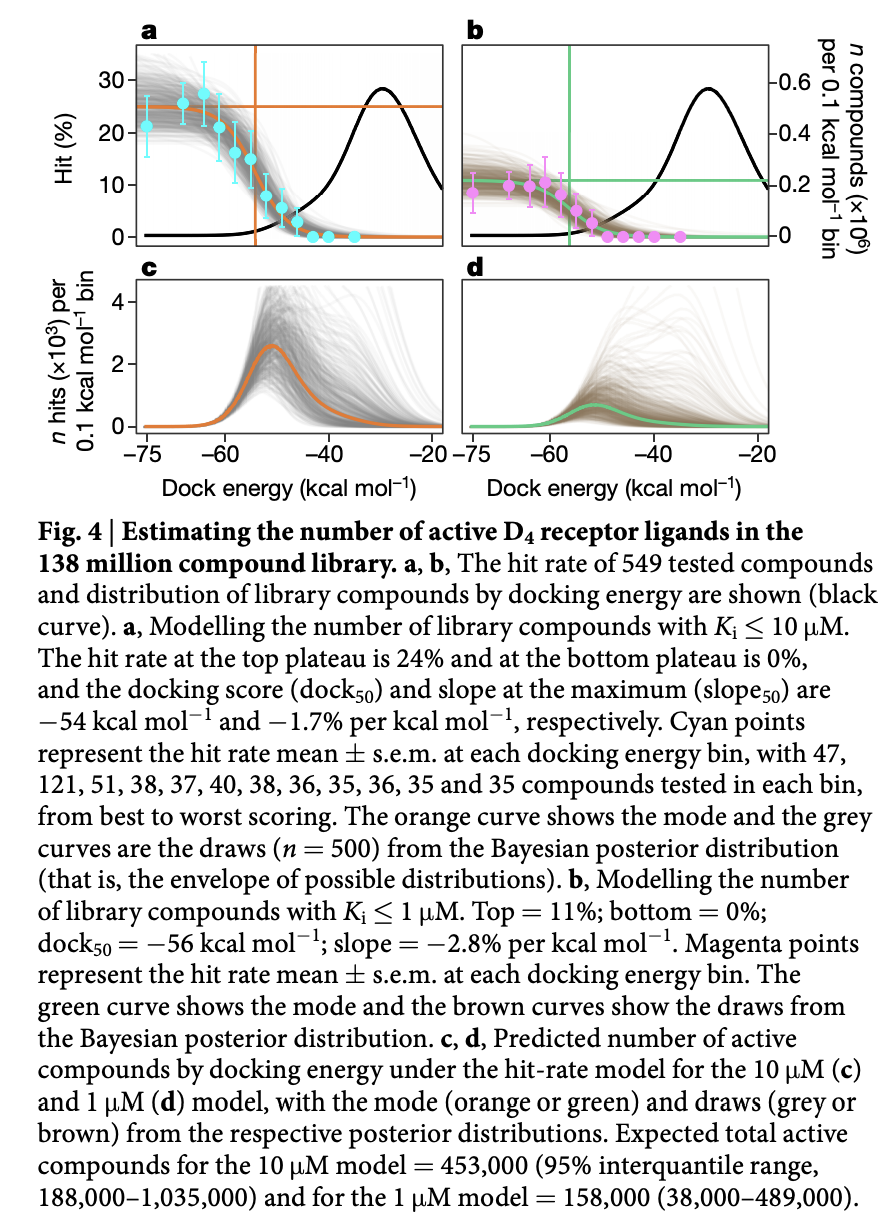
The appendix of this paper is worth a read to understand some of the details. Molecular docking is very dependent on proper data preparation and configuration, so the details are important. Protonation states and tautomers for compounds were done with chemaxon. As a quick reminder, tautomers are two forms of a molecule with different bond structures between which rapid interconversions happen. Here’s some examples:

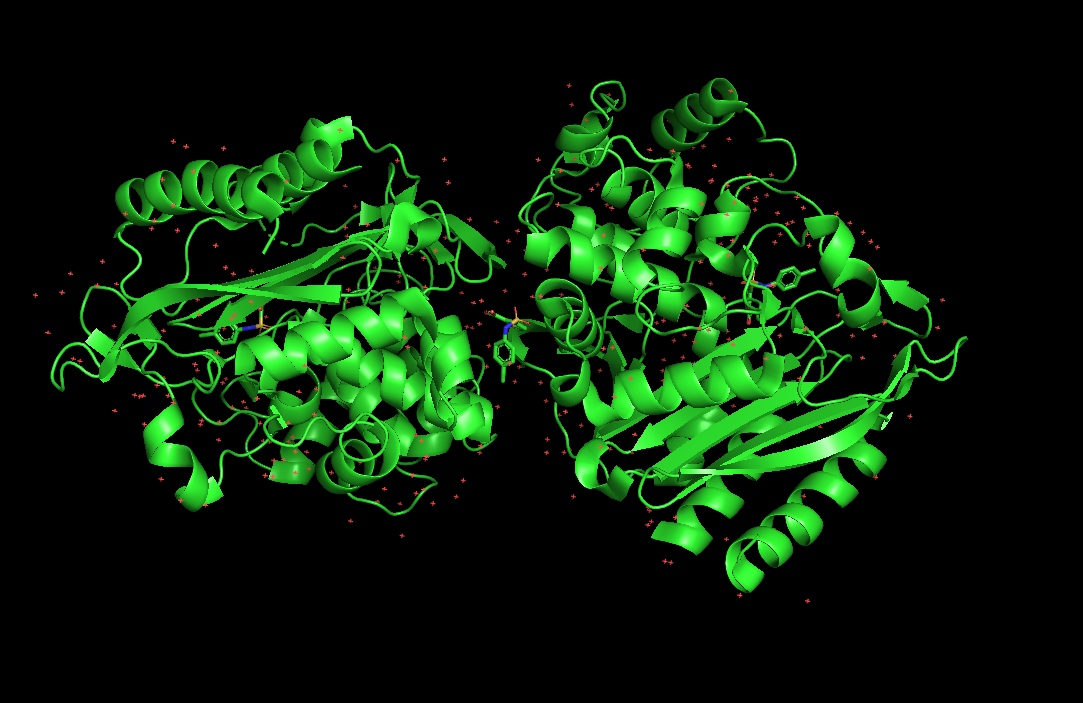
The AmpC campaign used structure in PDB 1L2S. Here’s a quick look at this structure using PyMol:
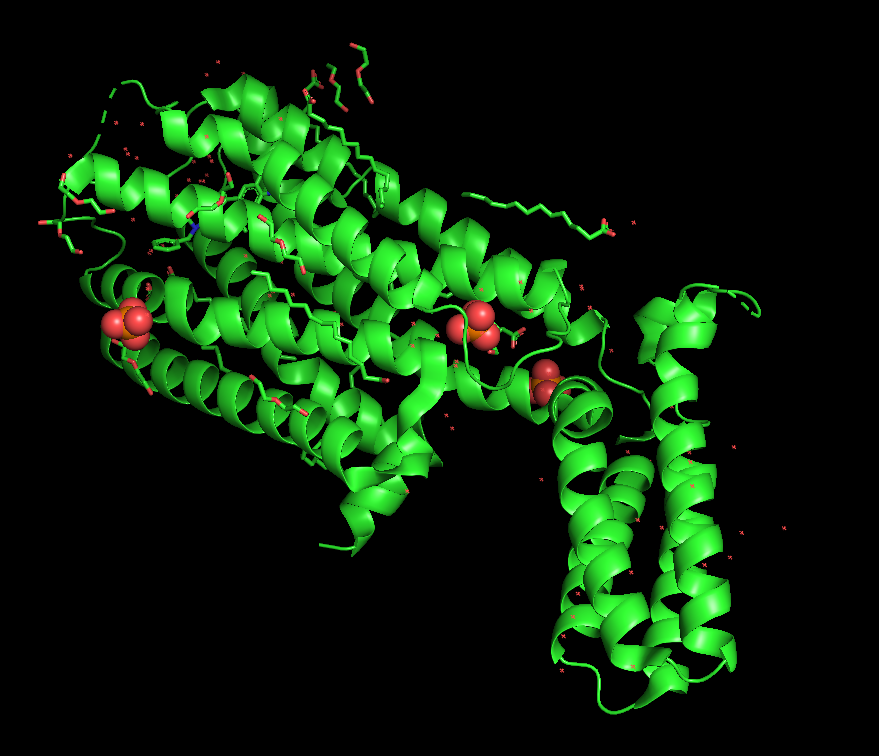
The PDB 5WIU was used for the \textrm{D}_4 Dopamine recepter. Here’s a quick look at that structure:
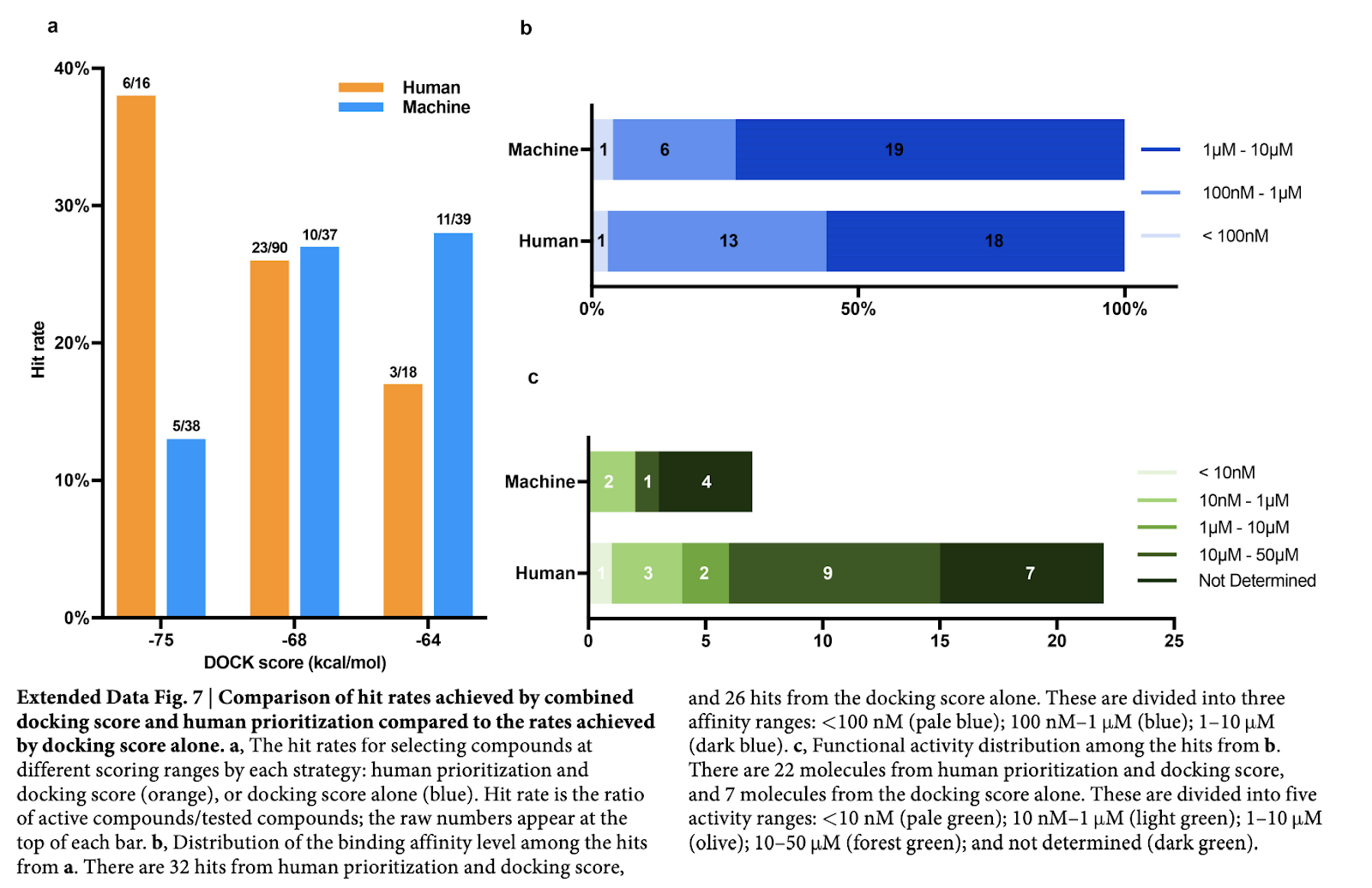
One final really interesting bit of analysis the authors did was comparing the docking algorithm’s performance to that of a human expert. Interestingly, it looks like when the docking score range was low, humans are much better at picking active compounds, but as the score range improves, the models outpace the humans.