
Preface
This summer I had the opportunity to contribute to an open-source codebase called DeepChem. I learned about DeepChem through the founder’s O’Reilley book: “Deep Learning for the Life Sciences.” As a computational biologist working in a genetics lab, I knew gaining experience with deep learning would be incredibly useful. I was prepared for a period of self-study but after joining the DeepChem developer calls, the energy and camaraderie of the community convinced me to apply for Google Summer of Code. My initial proposal was to integrate a genomic model into DeepChem and expand DeepChem’s support for genomic analysis. Looking back, I think it was very ambitious and I am glad I focused on the latter first.
DeepChem’s core functionality is small molecule chemistry but it has successfully branched out to materials science, quantum, and differential equations, as well as bioinformatics. This last branch has ample potential for development, especially on the topic of genomics. Prior to this project, DeepChem offered limited utilities for genomics and evaluation metrics for genomics datasets. There were only two bioinformatics tutorials available on DeepChem: one on an introduction to bioinformatics and the other on multiple sequence alignment (MSA). DeepChem also supported loading of FASTA files but could not handle any of the other numerous file formats used in genomics. These are a subset of the limitations that I tackled during my project this summer.
I hope to see DeepChem’s userbase expand as a consequence of the valuable contributions that my GSoC peers and I have made. This codebase is a powerhouse not only because of its utility but also because of its incredible mentors. If you are serious about gaining fluency in deep learning for the life sciences, DeepChem is the way to do it.
Special thanks to my mentor, Stanley Bishop, without whom this work would not have been possible.
Outline of contributions:
- Tutorials for DeepBio
- scVI tutorial
- ScanPy
- Bioinformatics files handling
- FASTQ
- VCF
- Additional contributions
Tutorials for DeepBio
-
scVI tutorial
- Recordings at single-cell resolution can give us a better understanding of the biological differences in our sample. As sequencing technologies and instruments have become better and cheaper, generating single-cell data is becoming more popular. In order to derive meaningful biological insights, it is important to select reliable analysis tools such as the one we will cover in this tutorial. scvi-tools (single-cell variational inference tools) is a package for probabilistic modeling and analysis of single-cell omics data, built on top of PyTorch and AnnData that aims to address some of the limitations that arise when developing and implementing probabilistic models. scvi-tools is used in tandem with Scanpy for which Deepchem also offers a tutorial. In the broader analysis pipeline, scVI sits downstream of initial quality control (QC)-driven preprocessing and generates outputs that may be further interpreted via general single-cell analysis tools.
- PR here
- Recordings at single-cell resolution can give us a better understanding of the biological differences in our sample. As sequencing technologies and instruments have become better and cheaper, generating single-cell data is becoming more popular. In order to derive meaningful biological insights, it is important to select reliable analysis tools such as the one we will cover in this tutorial. scvi-tools (single-cell variational inference tools) is a package for probabilistic modeling and analysis of single-cell omics data, built on top of PyTorch and AnnData that aims to address some of the limitations that arise when developing and implementing probabilistic models. scvi-tools is used in tandem with Scanpy for which Deepchem also offers a tutorial. In the broader analysis pipeline, scVI sits downstream of initial quality control (QC)-driven preprocessing and generates outputs that may be further interpreted via general single-cell analysis tools.
-
ScanPy tutorial
-
ScanPy is a scalable toolkit for analyzing single-cell gene expression data. It includes methods for preprocessing, visualization, clustering, pseudo-time and trajectory inference, differential expression testing, and simulation of gene regulatory networks. There are many advantages of using a Python-based platform to process scRNA-seq data including increased processing efficiency and running speed as well as seamless integration with machine learning frameworks.
- PR here
-
ScanPy is a scalable toolkit for analyzing single-cell gene expression data. It includes methods for preprocessing, visualization, clustering, pseudo-time and trajectory inference, differential expression testing, and simulation of gene regulatory networks. There are many advantages of using a Python-based platform to process scRNA-seq data including increased processing efficiency and running speed as well as seamless integration with machine learning frameworks.
Bioinformatics files handling
While doing user research, I found a big painpoint in the process of building models is processing the bespoke bioinformatic files. DeepChem already supports loading of FASTA files but not FASTQ or VCF. To do this I had to understand how to process and featurize genomic data and familiarize with what is already available in DeepChem. I also did research on what tools were available externally because I knew it was important not to reinvent the wheel and find an actual unmet need. Additionally, this required I figured out how to use loaders to translate genomics data into numerical representations that machine learning models can understand.
See below for more information (provided by Illumina) on what FASTQ files are and how they look like:
For each cluster that passes filter, a single sequence is written to the corresponding sample’s R1 FASTQ file, and, for a paired-end run, a single sequence is also written to the sample’s R2 FASTQ file. Each entry in a FASTQ files consists of 4 lines:
- A sequence identifier with information about the sequencing run and the cluster. The exact contents of this line vary by based on the BCL to FASTQ conversion software used.
- The sequence (the base calls; A, C, T, G and N).
- A separator, which is simply a plus (+) sign.
- The base call quality scores. These are Phred +33 encoded, using ASCII characters to represent the numerical quality scores.
Here is an example of a single entry in a R1 FASTQ file:
![]()
More detailed information on the FASTQ sequence file format can be found here.
See below for more information on VCF files (provided by GenomOncology):
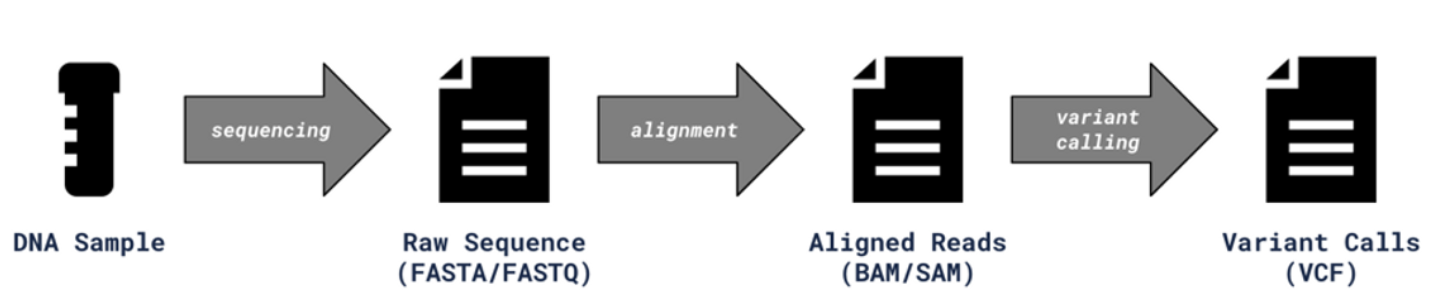
A variant call format file (VCF file) is the output of a bioinformatics pipeline. It specifies the format of a text file used in bioinformatics for storing gene sequence variations. Typically, a DNA sample is sequenced through a next generation sequencing system (NGS system), producing a raw sequence file. That raw sequence data is then aligned, creating BAM/SAM files as a result. From there, variant calling identifies changes to a particular genome as compared to the reference genome. That output is stored in a variant call format, VCF for short.
This is what a VCF usually looks like:

And below is a diagram of a typical bioinformatics sequencing pipeline.
- Read about how VCF files are used in machine learning and what features are important to extract
- More info here. Special thanks to Nico Alavi, authour of the post for meeting with me and providing guidance.
- Created a VCF loader with sharding enabled Unmerged PR
- Created a VCF loader that inherits from MolnetLoader to add VCF files into MolNet for benchmarking Unmerged PR
- Note: Both VCF loaders are unmerged because they imported
scikit-alleland we realized we did not want to have a dependency on a package that is not being actively mantained.
- Note: Both VCF loaders are unmerged because they imported
- Created a FASTQ loader with sharding enabled merged PR
Additional Contributions
- BibTeX citations for tutorials
- See here for PR
- Some PRs still under review:
- Researched a good initial dataset to add to DeepBio:
- Data from Bone Marrow Mononuclear Cells (BMMCs) would be a great candidate due to the reasons outlined in this video (time stamp 14:20) outlined below and in the picture below.
- The system is well understood
- We know cell types present in the bone marrow and what surface markers are expressed.
- Mixture/Variety of different cell types (both spectrum and clusters)
- Commercially available
- https://github.com/deepchem/deepbio/issues/2
- Data from Bone Marrow Mononuclear Cells (BMMCs) would be a great candidate due to the reasons outlined in this video (time stamp 14:20) outlined below and in the picture below.
- Diagram for Hugging Face and DeepChem integration. See here
- Read about how data structure objects are handled in both HF and DC to start thinking about the bridge between fitting and training models in these two ecosystems. Rough notes here.
- Enformer presentation. See here
- A neural network architecture developed by a Calico + DeepMind team which uses transformers to incorporate long-range sequences for gene expression prediction.
- Lindau Meeting on AI for Chemistry and Biology
- I got to showcase and promote DeepChem to more than 700 young scientists from around the world. Read more
- Learn about DeepChem’s best practices for version control, testing, code review, and communication. Here is a useful chart I created for convention checking:
| yapf 0.32 | code formatting |
|---|---|
| flake8 | linting code syntax, stylistically |
| doctest | docstrings |
| pytest | functionality- arguably the most important. Also runs unittest|
|
| mypy | static type checker |
yapf -i <modified file>-
flake8 <modified file> --count
* python -m doctest <modified file> pytest path/to/file -vmypy --follow-imports=skip path/to/file
Future Plans
I am currently looking at the metagenome pipelines from New Atlantis as a motivational case for a DeepChem-ic assembly class. If you are interested in reading more about what Russian bridges, Eulerian circuits, and genome assembly have in common I recommend this blog post. Recent advances leveraging neural architectures for genome assembly such as this one are very exciting and could certainly be more easily implemented by implementing them in a modular fashion.