
Final Submission of MXMNet Model Implementation for DeepChem

I had the privilege to participate in Google Summer of Code 2023 as a contributor. During this time, I collaborated with DeepChem (Open Chemistry), a project that aims to create high-quality, open-source tools to democratize the use of deep learning in drug discovery, materials science, quantum chemistry, and biology.
Among its suite of various machine learning models, it has a wide range of graphs-based neural network model implementations aimed to solve the applications such as predicting the solubility of small drug-like molecules, binding affinity for a small molecule to protein targets, analyzing protein structures, and extracting useful descriptors.
In this blog post, I intend to provide a comprehensive documentation and detailed description of all the tasks I accomplished throughout the summer.
Project Title
MXMNet Model Implementation for DeepChem
This project seeks to bring a new tool to the DeepChem suite for solving message passing problems based on the recent advancements in the GNNs research. The project aims to implement a Multiplex Molecular Graph Neural Network (MXMNet)[1] model to DeepChem.
Some Important Links
My GitHub: https://github.com/riya-singh28
DeepChem Home Page: https://deepchem.io/
DeepChem GitHub: https://github.com/deepchem/deepchem
Proposal Link: Link
Preface
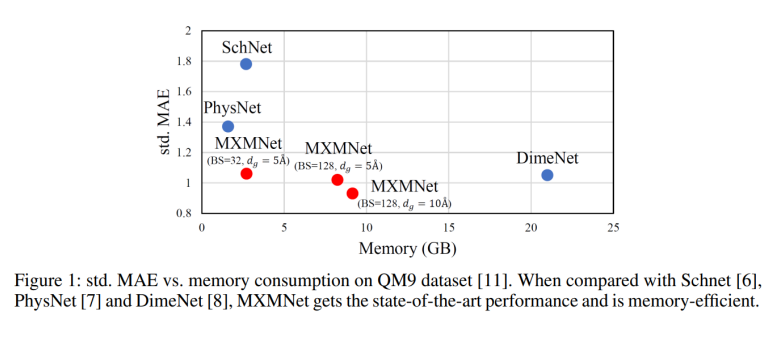
The primary difference between MXMNet and regular message passing networks is, previous GNNs have used auxiliary information such as chemical properties and pairwise distances between atoms, but this increases computational complexity. MXMNet uses angular information to model only local connections, avoiding expensive computations on all connections, and divides molecular interactions into two categories: local and global. It constructs a two-layer multiplex graph where one layer contains local connections and another layer contains global connections, and designs a Multiplex Molecular (MXM) module that performs efficient message passing on the whole multiplex graph. On both datasets, QM9 and PDBBind, the model can outperform the baseline models. Regarding the efficiency, the model requires significantly less memory than the previous state-of-the-art[1] as shown in Figure 1 and achieves a training speedup of 260%.
Contents covered in the blog
– Expected Deliverables
– Featurization of molecule SMILES
– Overview of Architecture of MXMNet Model
– MXMNet Workflow
– Fixes and Additions to DeepChem
– Future Scope
– Acknowledgement
– References
Expected Deliverables
Implementation of classes and support functions for featurization of SMILES data.
Implementation of model framework for MXMNet. (In Progress)
Featurization of molecule SMILES
Cheminformatics datasets contain molecules represented as SMILES . These SMILES can be analysed using the RDKit library to get information about the atoms and bonds in the molecules.
Chemical structures and their respective SMILES
Molecular fingerprinting is a vectorized representation of molecules capturing precise details of atomic configurations. During the featurization process, the atomic numbers Z are represented with randomly initialized, trainable embeddings to be the input node embeddings through a graph data class. The graph data class also contains x, y, z coordinates value for each item in the molecule graph.
For this project, I implemented atomic level graph featurizer in DeepChem, specific to MXMNet model requirements.
Multiplex Graph
After Featurization, we proceed to generate interaction graphs that encapsulate diverse Geometric Information (GI).
- To create the local GI, we establish edges by either utilizing chemical bonds or identifying neighboring nodes within a short cutoff distance, a choice contingent upon the specific investigative task.
- For the global GI, we form edges by defining neighbors for each node within a comparably larger cutoff distance.
These interaction graphs are then treated as layers to construct a multiplex molecular graph denoted as G = {Gl, Gg}, comprising a local layer, Gl, and a global layer, Gg. This resultant G is subsequently employed as the input for our model.
Corresponding PRs:
Overview of Architecture of MXMNet Model
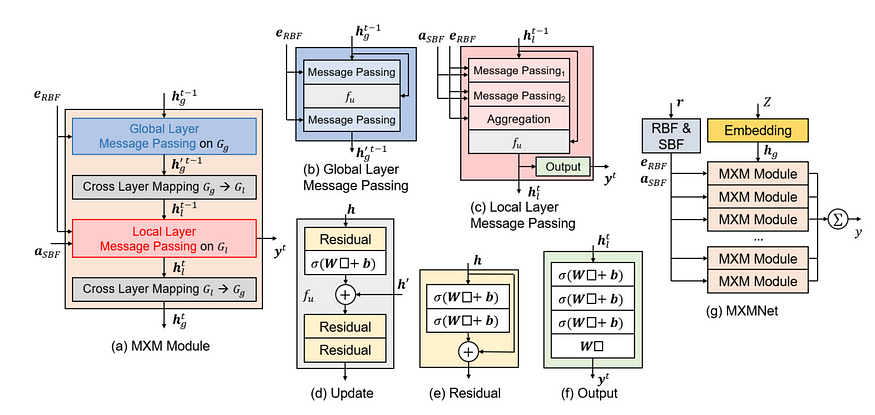
Overview of the architecture of the MXM module and the MXMNet. In the illustrations, σ denotes the non-linear transformation, denotes the input for the layer.
MXMNet Workflow
MXMNet implementation works in 2 phases:
● Message passing phase
● Readout phase
Message Passing Modules:
Global layer message passing module consists of two identical message passing operations that can capture the pairwise distance information. Each message passing operation is formulated as follows:
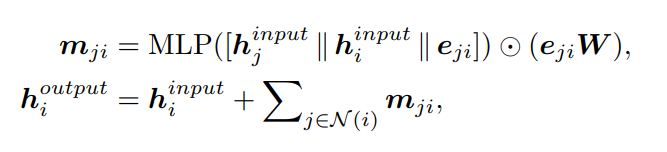
where i, j ∈ G(global) , the superscripts denote the state of h in the operation. In our global layer message passing, an update function fu is used between the two message passing operations. We define fu using multiple residual modules. Each residual module consists of a two-layer MLP and a skip connection. The resulting operation will only need O(2Nk) messages.
Local Layer Message Passing module utilizes both pairwise distance and angles to represent local interactions. There are two types of angles associated with the edges: two-hop angles between the one-hop edges and the two-hop edges (∠ij1k1 , ∠ij1k2 ), and one-hop angles that are formed solely by the one-hop edges (∠j1 ij2 and ∠j1 ij3 ) .
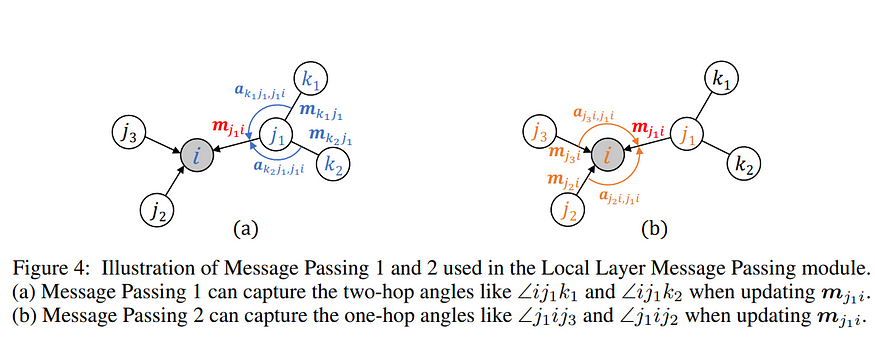
Step 1: Message Passing 1 — captures the two-hop angles and related pairwise distances to update edge-level embeddings {mji} .
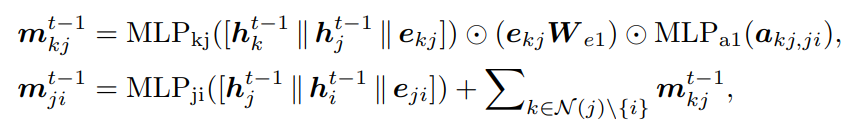
Step 2: Message Passing 2 — captures the one-hop angles and related pairwise distances to further update {mji} .
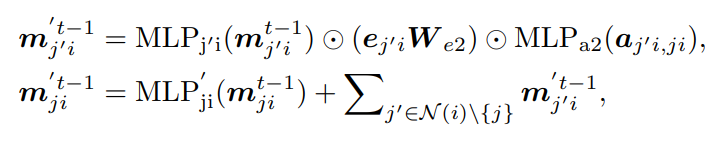
Step 3: Aggregation and Update — aggregates {mji} to update the node-level embedding (hi)
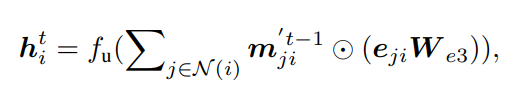
where i, j, k ∈ Glocal , a(kj,ji) is the feature for angle α(kj,ji ) = ∠h(k)h(j)h(i ). We define fu using the same form as in the global layer message passing. These steps need O(2Nk^2 + Nk) messages in total.
Cross Layer Mapping function f(cross) takes either the node embeddings {hg} in the global layer or the node embeddings {hl} in the local layer as input, and maps them to replace the node embeddings in the other layer

where g ∈ Gglobal, l ∈ Glocal, the fcross and f’cross are learnable functions. In practice, we use multi-layer perceptrons to be fcross and f’cross . Each of them needs O(N) messages being updated.
In the RBF & SBF module, the Cartesian coordinates r of atoms are used to compute the pairwise distances and angles. We use the basis functions proposed in [2] to construct the representations of eRBF and aSBF .
Readout Phase
This phase makes use of the pool.global_add_pool method. It adds node features across the node dimension, and returns batch wise graph-level-outputs, so that for a single graph its output is computed by
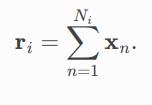
Corresponding PRs:-
- Global Message Passing Layer for MXMNet Model.
- Local Message Passing Layer for MXMNet Model.
- MXMNet Envelope Layer
- RBF Module- MXMNetBesselBasisLayer
- SBF Module- MXMNetSphericalBasisLayer
Then we stack MXM modules to perform message passings. In each MXM module, we use an Output module to get the node-level output. The final prediction y is computed by summing all outputs together among all nodes and all layers.
Corresponding PRs for Model:-
Fixes and Additions to DeepChem
I have made a total of 13 PRs, 63+ commits, and 1920+ additions during this summer .
DeepChem suite now includes:
- A fully functional SMILES featurizer for the MXMNet model with support for molecular features.
- Pytorch Utilities — Unsorted segment sum and Segment Sum functions.
- Egdenetwork Layer ported from Tensorflow to Pytorch.
- Changes in MultilayerPerceptron to work as a ResidualBlock as well by passing a weighted_skip parameter.
- Envelope layer, Bessel Basis, Spherical Basis, Global and Local Message Passing layer for MXMNet Model.
In progress:
- A new torch model class for MXMNet, with support for batching.
Next, I plan to add a tutorial to use the MXMNet model.
You can find my progress reports, made throughout the summer, for the project at the following DeepChem forum link:
Future Scope
In the future, there is potential for MXMNet model implementation to include benchmarking against various regression-based datasets available in Molnet. It would also be exciting to explore the classification capabilities of the MXMNet model by making a few architectural changes to adapt it for classification-type datasets.
Acknowledgment
I express immense gratitude for all the support extended by my mentors Bharath Ramsundar , Vignesh , Aryan Amit Barsainyan and the DeepChem community for their suggestions and discussions.
I’m thrilled to be part of an organization where I can gain extensive insights into the practical aspects of ML models within advanced expert systems, with a focus on graph-based neural networks. My plan is to make DeepChem even better by fixing things, making improvements, and helping out the community whenever I can.
Thank you, Google, for this fantastic opportunity.
References
[1] Molecular Mechanics-Driven Graph Neural Network with Multiplex Graph for Molecular Structures