
I had the opportunity to be a Google Summer of Code student working with DeepChem, an open-source organization democratizing deep learning for chemistry. The repository contains a suite of models , featurizers and datasets from literature and other sources, allowing chemistry-oriented and other interested practitioners to build state-of-the-art models for chemistry applications. For more details into how DeepChem’s API is structured, please refer to Wu et. al [2].
In this post, I intend to describe the work I carried over the summer, motivating and explaining necessary concepts along the way.
The title of my project was A Transfer Learning Framework for DeepChem, and expected deliverables include:
- Testing a Transfer Learning protocol described in Goh et al [1] and verify reproducibility of results
- Putting together an API, a blogpost and for training a model and fine-tuning it on a DeepChem dataset and compare results
With chemistry becoming a more data-driven discipline in recent years, deep learning models have found applications in various molecular and quantum property prediction tasks, even reaching state-of-the-art in some of these. However, the amount of data available for training deep learning models is significantly smaller than their imaging counterparts (ImageNet, for example), lending them prone to overfitting and also hampering our ability to build deeper, more expressive models.
Two common strategies for dealing with limited training data include:
-
Gathering more training data
This is difficult in chemistry for two reasons:
- Experiments demand considerable time and effort and are not fully automated
- Material and reagents are available in small quantities and are expensive hence experiments need to be targeted
-
Training the model on a “similar” but large dataset which is already labelled, or for which proxy labels are easy to compute. Once the model is trained on the larger dataset, it is then fine-tuned on the smaller dataset of interest. This idea is called Transfer Learning.
The second idea is the focus for the rest of the blog. The blog is organized as:
- Introduction
- ChemNet - Transfer Learning Protocol for Chemistry
- SmilesToSeq and SmilesToImage Featurizers
- ChemCeption and Smiles2Vec models
- Fixes and Additions to DeepChem
- Transfer Learning Protocol
Transfer Learning - Introduction
Human learners appear to have inherent ways to transfer knowledge between tasks, ie) they can take previous learning experiences and apply them when encountering a new task. The more related the new task is to previous experience, the more easily they can master it.
Common machine learning algorithms consider only isolated tasks and performance improvement on those tasks. Transfer learning aims to address this by developing methods that train models first on a set of source tasks, then use the trained model to improve learning on the target task. Techniques that facilitate knowledge transfer between tasks aim to make machine learning as efficient as human learning.
Performance improvements from transfer learning can be investigated using three measures:
- Initial performance using only the transferred knowledge, without any further learning on the dataset pertaining to the task of interest
- Time taken for learning the new task with the transferred knowledge as opposed to learning from scratch
- Final performance using transferred knowledge as opposed to training from scratch
This type of transfer learning is also known as inductive transfer, as the model learns inductive biases by learning a set of source tasks and then uses those biases to inform its predictions on a target task.
ChemNet - Transfer Learning Protocol in Chemistry
Goh et. al [1] describes a weak supervised learning based protocol called ChemNet to carry out inductive transfer, that is model and dataset agnostic. To support this claim, the authors evaluate the performance on three datasets from MoleculeNet[2] (Tox21, HIV, Sampl)using two different models, ChemCeption and Smiles2Vec and find an improvement in performance across all the datasets using the protocol. The following sections describe ChemNet, the featurization routines, the training procedures and the two models discussed in this paper.
The protocol, in its essence is straightforward - Starting with a large dataset of unlabelled molecules, several molecular descriptors are computed approximately using the chemoinformatics package RDKit. The authors used the ChEMBL dataset for the molecules, which in its latest version (25) has around 1.8 million molecules. The number reduces by a few thousand when the SMILES strings are canonicalized and redundancies are eliminated. For the molecules in the dataset, the authors compute 100 molecular descriptors under categories:
- Topological / Topochemical Properties - BalabanJ, BertzCT
- Surface Area descriptors
- Atomic and Substructure compositions - Number of aromatic rings, heteratoms, NH-OH content
The pretraining task is an instance of multitask learning allowing the model to learn atomic and substructural compositions of the molecule and how they relate to other higher level properties as mentioned above.
Features are computed on molecules compatible with the downstream models to be used, and the (featurized molecules, descriptors) tuple becomes the dataset. The dataset is divided into training, validation and test datasets, with a 4:1:1 split ratio. The output values were transformed using the MinMax transformer to scale all values between 0 and 1. Models were trained for 50 epochs to minimize the L2Loss, with an early stopping criterion monitoring the validation loss every 10 epochs.
Finetuning was carried out by loading part of the pretrained model, except the final layer. The entire model was trained for 500 epochs with an early stopping criterion monitoring the validation loss every 50 epochs. For the classification tasks, BinaryCrossEntropy loss was used, while L2Loss was used for the regression tasks. The authors perform the finetuning on Tox21 (classification), HIV(classification) and FreeSolv(regression) datasets. I carry out the training on more datasets from MoleculeNet which include SIDER (classification), Clintox (classification), Delaney (regression), Toxcast(classification) datasets. The results for the same can be found in the Transfer Learning section.
For both pretraining and finetuning, RMSProp optimizer was used with its standard settings. The paper mentions oversampling to handle class imbalance. Class imbalance is handled in DeepChem by assigning different weights to classes based on their frequency and is accounted for when the losses are calculated. This difference between the original paper and DeepChem’s implementation will cause differences in the final performance, although it is not expected to be too significant.
Dataset download and Preparation
My first task was downloading and preparing the dataset. The dataset (in sdf format) can be found here. The code for preparing the dataset can be found under this repository.
As the dataset has around 1.8 million molecules with 100 descriptors to be computed for each molecule, I used the multiprocessing package to utilize multiple cores for faster processing. Intermediate results for every 10000 molecules processed were saved in addition to the final results. This final processed dataset was hosted on DeepChem’s AWS server, found here. Preprocessing was a preparatory task and not the main focus of this project, so I refrain from detailing the process and encourage those interested to look at the repository.
The next step was writing a loader function in a style similar to rest of loading functions in DeepChem. A loader function takes some arguments regarding the directories to store and download data to, the type of split and featurizers to be used and optionally, other dataset specific arguments. The loader function written for the ChEMBL25 dataset can be found here. The code below shows how to setup the featurized dataset using DeepChem:
import deepchem as dc
# tasks is a list of task names associated with the dataset
# dataset is a three element tuple of train, valid and test datasets
# transformers is a list of transformers applied on the dataset
tasks, dataset, transformers = dc.molnet.load_chembl25(data_dir=None, save_dir=None,
featurizer='smiles2img', split='random')
This will download the processed file to a data directory (temporary in this case), featurize shards of molecules, split the datasets into training, validation and testing and save it to disk. The following table shows the featurizer and split options available for this loader:
| Featurizers | Splits |
|---|---|
| ECFP (Extended Connectivity Fingerprint) | Random |
| GraphConv (Turn Molecule into graph and prepare relevant data structures) | Index |
| SmilesToImage (Turn SMILES based representations into images) | Scaffold |
| SmilesToSeq(Turns SMILES based representations into sequences) | |
| Weave (Featurizer for WeaveModel) | |
| Raw |
SmilesToImage and SmilesToSeq featurizers
Principle
SmilesToImage featurizer takes a SMILES string as an input and turns it into an image based representation. The authors use two types of image representations - a single channel representation (called “std”) and a four channel representation (called “engd”).
In the “std” representation, the 2D coordinates of the atoms in the molecule are first computed and scaled according to image size. The pixel locations corresponding to atoms are filled with their atomic numbers. Bonds are represented by straight-lines drawn between the corresponding atoms and the pixel locations are filled with the value 2 (which does not correspond to the identity of any element). In the “engd” representation, the first channel follows the same methodology as the “std” representation. The remaining three channels are applicable only to the atom pixel locations and correspond to properties like hybridization, partial charge and valency.
The other featurizer used is SmilesToSeq, which takes a SMILES string as input and turns it into a sequence of integers depending on the characters in the SMILES string.
Implementation and Usage
Both SmilesToImage and SmilesToSeq are derived from DeepChem’s Featurizer class (deepchem.feat.Featurizer). SmilesToSeq takes as input a character to index mapping, the maximum sequence length to process and padding to add to either side of the sequence. An example usage would be:
from deepchem.feat import SmilesToSeq
from rdkit import Chem
char_to_idx = {'C': 1, 'O': 2}
max_len = 20
pad_len = 5
featurizer = SmilesToSeq(char_to_idx=char_to_idx, max_len=max_len, pad_len=pad_len)
example_smiles = ['CCCCO', 'CCC']
example_mols = [Chem.MolFromSmiles(smile) for smile in example_smiles]
featurized_smiles = featurizer.featurize(example_mols)
There are additional utilities to transform the sequences back to SMILES, which can be used if needed.
SmilesToImage takes as input arguments the image size, resolution of each pixel and maximum sequence length. The resolution is a value in Angstrom, which are the usual units for atomic distances and controls the scaling. An example usage would be:
from deepchem.feat import SmilesToImage
from rdkit import Chem
max_len = 20
featurizer = SmilesToImage(img_size=80, max_len=max_len, res=0.5)
example_smiles = ['CCCCO', 'CCC']
example_mols = [Chem.MolFromSmiles(smile) for smile in example_smiles]
featurized_smiles = featurizer.featurize(example_mols)
The corresponding code for both the featurizers can be found here.
Pull Requests:
#1618: Smiles Based featurizers for ChemNet
ChemCeption and Smiles2Vec models
The authors describe two models in the paper, ChemCeption and Smiles2Vec. ChemCeption takes as input the image based representations of molecules while Smiles2Vec operates on sequences. The SmilesToImage and SmilesToSeq featurizers are used before feeding the inputs to the respective models.
ChemCeption
ChemCeption is based on the Inception-Resnet v2 model from Google and follows the same architecture, except for a longer Stem segment in the original model. The model uses specially designed Inception-ResNet layers, which allow the feature maps to be processed at multiple scales in the same layer (the Inception portion) in addition to adding the information from the original feature map (ResNet connection). They also use Reduction layers which reduce the spatial dimension of the feature maps to control computational complexity. A simplified diagram of the architecture:
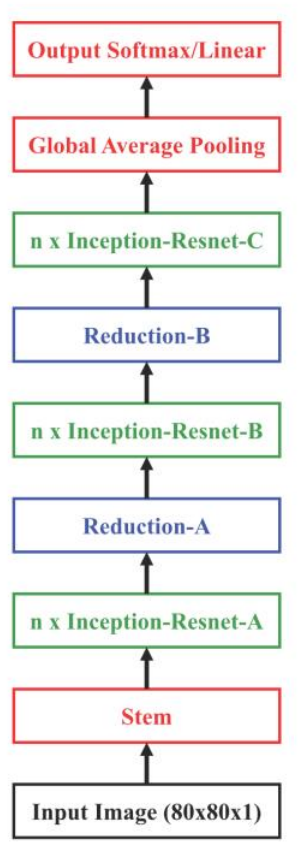
The different layers in the model are designed as:
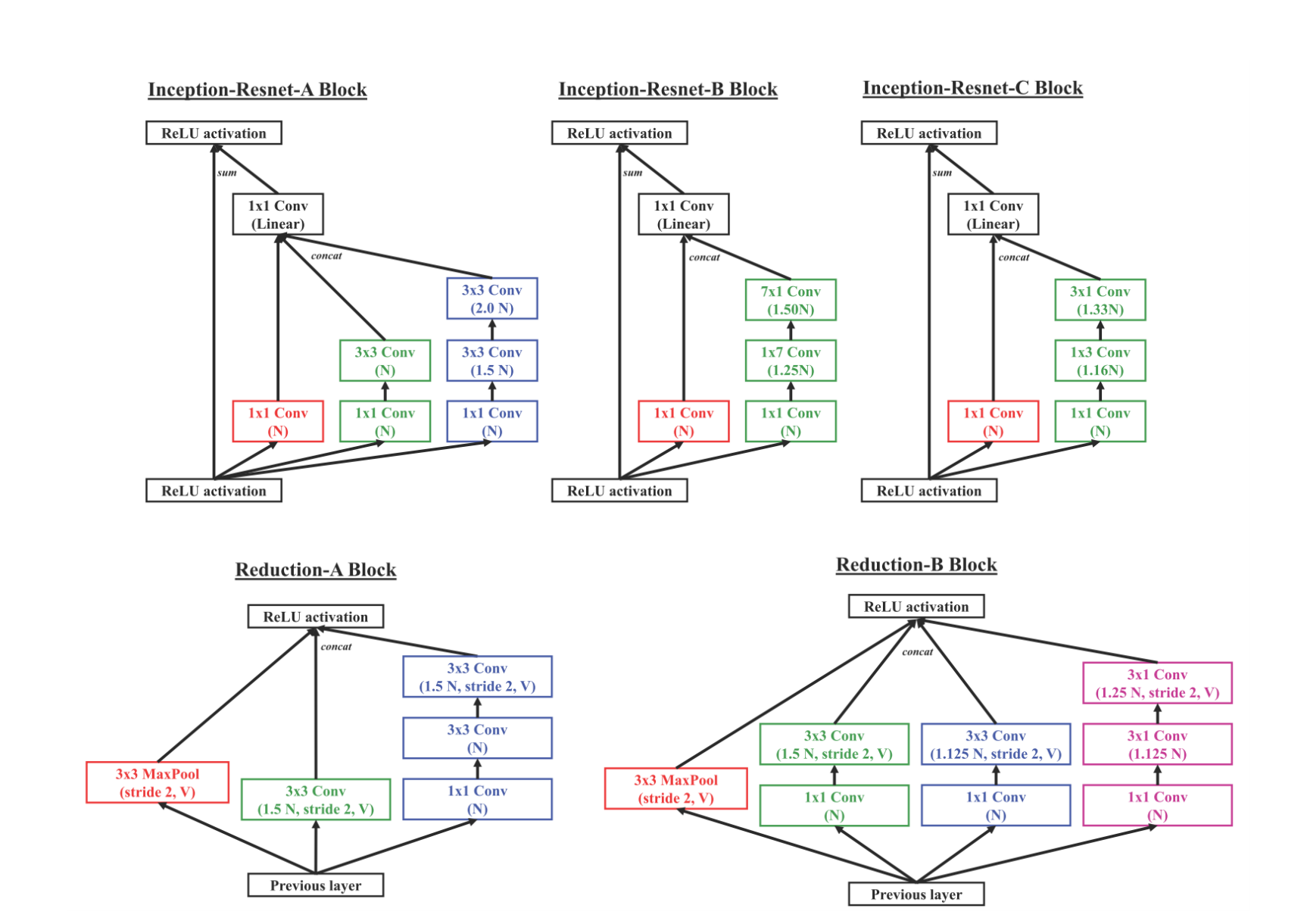
N indicates the number of base filters used in the model & V indicates valid padding.
Smiles2Vec
Smiles2Vec has a simple architecture: an embedding layer (character level) followed by an optional Convolutional layer, two layered bidirectional RNN (GRU or LSTM cells) followed a final Softmax or linear layer depending on a classification or regression task.
Implementation and Usage
Both models were implemented as subclasses of DeepChem’s KerasModel(deepchem.models.KerasModel), which is a wrapper around tf.keras.Model allowing interoperability with DeepChem’s datasets. Implementations of the ChemCeption and Smiles2Vec models can be found here. The Inception-Resnet layers and ResNet were implemented as subclasses of tf.keras.layers.Layer and can be found here.
Pull Requests:
#1623: Added ChemNet models with tests
An example usage of ChemCeption model would be:
import deepchem as dc
tasks, dataset, transformers = dc.molnet.load_chembl25(featurizer='smiles2img', split='random', img_spec='std')
train, valid, test = dataset
model = dc.models.ChemCeption(img_spec='std', n_tasks=len(tasks), mode='regression')
model.fit(train, nb_epoch=10)
Fixes and Additions to DeepChem
In addition to preparing the setup for Transfer Learning, I also had to make some additions and fixes to the existing code inside deepchem.molnet.
-
The first fix that was added was to apply the transformers to the datasets only after splitting and not before. This fix was applied to a major fraction of datasets within MoleculeNet.
Pull Requests
#1621: Swap Split-Transform order - I
#1624: Swap Split-Transform order - II -
Addition of MinMax Transformer into
deepchem.transalong with corresponding tests and documentation for the same. Apart from that, the pull request also added split fraction options for the Tox21, HIV and FreeSolv datasets (these datasets were used for evaluation in the paper), along with the option of saving and downloading to custom directories.Pull Requests
#1638: ChemNet Fixes and additions
-
Added SmilesToImage featurizers for all datasets inside MolNet, along with options for custom directories for downloading and saving datasets to.
Pull Requests
#1637: SmilesToImage featurizers for Tox21, HIV and Sampl Datasets
#1649: Custom directories and SmilesToImage for MolNet
#1651: ChemNet Fixes
#1660: Stratified splitters and minor changes for MolNet -
Upgrade DeepChem to TF 1.14
Checkpoint saving for nested layers (Inception-Resnet layers in our case) was enabled by default only in TF 1.14. Hence the upgrade had to be carried out for saving trained ChemCeption models correctly.Pull Requests
#1645: Upgrade to TF 1.14
Transfer Learning
The script used in the transfer learning protocols can be found here.
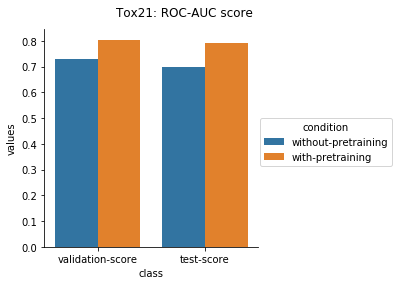
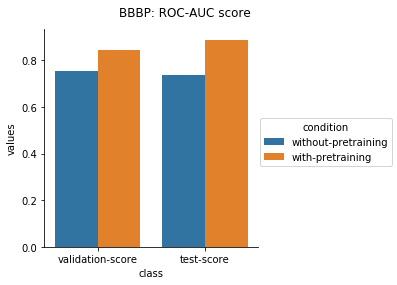
The results from applying the transfer learning protocol described above are shown below.
Physiology Datasets in MoleculeNet
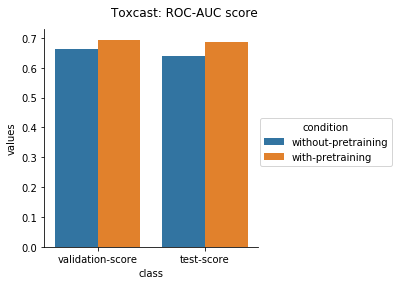
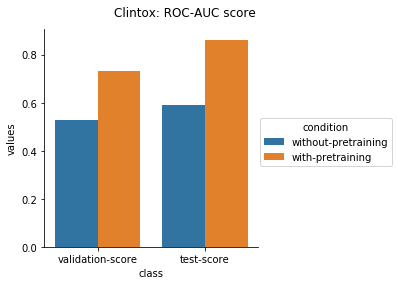
Physical Chemistry Datasets
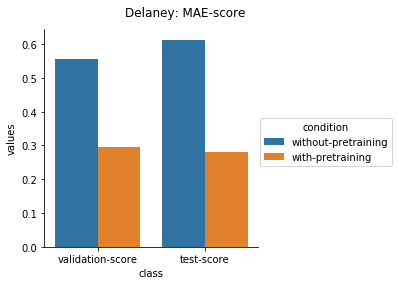
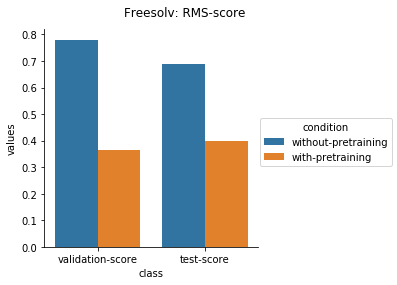
Quantum Mechanical Datasets
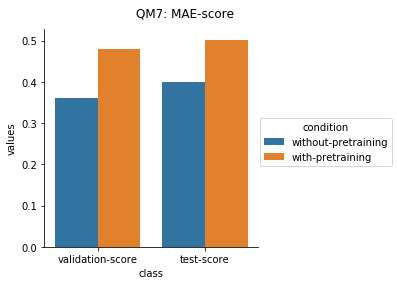
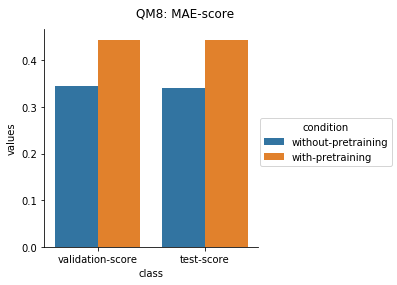
In the case of Toxcast datasets, the improvements look small because the model is learning to predict the outcome on 617 tasks. For the other datasets where the predictions are on fewer tasks, the results are much better.
For the quantum mechanical datasets, the results are interesting - negative transfer is observed. I reckon this is because the pretraining step involves prediction on topological and physicochemical properties and not on quantum chemistry based properties. The finetuning stage ends up using the dataset to unlearn parts of the previous pretraining step and then start relearning representations to better predict quantum chemical properties.
Implementation & Usage
Loading the pretrained model
Having an API that allows a user to load parts or whole of a pretrained model was the central goal of the project. Loading from a pretrained model involves copying respective weight values to another model (that shares some of its architecture with the pretrained version), which is what our API tries to reflect. The workhorse is a function load_from_pretrained, the PR for which can be found here.
The main arguments of the function are:
-
source_model: Pretrained model or a copy with the same architecture -
assignment_map: Dictionary where key-value pairs are the variables in the pretrained vs current model -
value_map: Values of different variables in the pretrained model. If this is unavailable, the model is restored from a checkpoint
One of the simple tasks to try transfer learning on is training the model on a classification or regression task and finetuning it on a smaller dataset for a different task mode or a different number of tasks within the same mode. Considering the network as an organized collection of layers, loading from a pretrained model is equivalent to copying over all layer variables except the final layer.
Many models inside DeepChem have this structure. To allow users to quickly get started with transfer learning for these tasks, if assignment_map is None, we create a default mapping assuming the structure in the previous paragraph. For the more advanced users who want to load or retrain certain segments of the model architecture, a custom assignment_map can also be defined.
Since a tf.keras.model can be defined without using any layers (while still having trainable variables), the assignment_map operates at the level of variables. Its a tradeoff that we made between using a more general API operating at a finer scale (variables) vs an API with an assumed graph structure that operates on a coarser scale (layers). Its a little tricky to get started with initially, especially for custom fine-tuning but tf.keras.Model adds variables in the same sequence as the layers are created, making it easier to create the assignment maps with shared correspondences between the pretrained and finetuning model.
Below is a simple example that indicates how to use this function.
A MLP class is defined and the model is trained on a toy dataset to predict 100 values per input. In the second case, only 10 values are predicted and the weights from the previous model are loaded except for the final layer.
import deepchem as dc
import numpy as np
import tensorflow as tf
from deepchem.models.losses import L2Loss
class MLP(dc.models.KerasModel):
def __init__(self, n_tasks=1, feature_dim=100, hidden_layer_size=64,
**kwargs):
self.feature_dim = feature_dim
self.hidden_layer_size = hidden_layer_size
self.n_tasks = n_tasks
model, loss, output_types = self._build_graph()
super(MLP, self).__init__(
model=model, loss=loss, output_types=output_types, **kwargs)
def _build_graph(self):
inputs = Input(dtype=tf.float32, shape=(self.feature_dim,), name="Input")
out1 = Dense(units=self.hidden_layer_size, activation='relu')(inputs)
final = Dense(units=self.n_tasks, activation='sigmoid')(out1)
outputs = [final]
output_types = ['prediction']
loss = dc.models.losses.BinaryCrossEntropy()
model = tf.keras.Model(inputs=[inputs], outputs=outputs)
return model, loss, output_types
X_1 = np.random.randn(100, 32)
y_1 = np.random.randn(100, 100)
dataset_1 = dc.data.NumpyDataset(X_1, y_1)
X_2 = np.random.randn(100, 32)
y_2 = np.random.randn(100, 10)
dataset_2 = dc.data.NumpyDataset(X_2, y_2)
source_model = MLP(feature_dim=32, hidden_layer_size=100, n_tasks=100)
source_model.fit(dataset_1, nb_epoch=100)
dest_model = MLP(feature_dim=32, hidden_layer_size=100, n-tasks=10)
dest_model.load_from_pretrained(
source_model=source_model,
assignment_map=None,
value_map=None,
model_dir=None,
include_top=False)
dest_model.fit(dataset_2, nb_epoch=100)
MoleculeNet oriented example can be found under the chemnet-auxillaries repository.
In addition to this, I have also put together a simple script to allow transfer learning between any model in DeepChem and a MoleculeNet dataset. The PR for this is not merged yet, and under review. In this steup, configurations for loading ChEMBL25 and a MoleculeNet dataset must be specified, in addition to the hyperparameters for the pretrained and finetune model architecture. run_finetuning operates under the assumption that the all layers except the final layer will be copied from the pretrained model. I will setup a Jupyter Notebook based tutorial soon, which will be referenced here.
Acknowledgments
A special thanks to Bharath and Peter for insightful discussions and advice throughout the summer. It was great fun working with you guys and I hope to continue contributing to DeepChem!
References
[1]: Using Rule-Based Labels for Weak Supervised Learning
[2]: MoleculeNet: A Benchmark for Molecular Machine Learning
 Anyway, thanks for the clarification!
Anyway, thanks for the clarification!