Over the last several years DeepChem has evolved from a very limited set of scripts (my first implementation could only train multitask networks on chemoinformatic data) into a sophisticated system for scientific machine learning. At the same time, DeepChem has a number of limitations that prevent it from addressing many important scientific problems. The core of DeepChem is rigid; it isn’t easy for users to compose DeepChem tools to make custom models and architectures without diving into the internals of DeepChem. DeepChem is also designed to run on single devices, so it’s hard to tackle large scale scientific problems. This note studies DeepChem’s current design and highlights awkward APIs, rigid constructions, and scaling issues. I then suggest improvements to DeepChem’s core to address these challenges.
I want DeepChem to evolve into a powerful framework for AI-driven science and engineering and enable users to solve hard problems in other fields of science (such as semiconductors, energy, climate and more) in addition to our core strengths in open source drug discovery. This transition will require us to generalize and expand DeepChem’s API for building sophisticated and scalable scientific programs.
The proposals here are first sketches which will require major work to turn into actual implementations. This write-up draws on a number of discussions that we’ve had at DeepChem developer calls on DeepChem flexibility, model pretraining, model hubs, and many more topics. The developer calls are a continuing source of inspiration and my sincere thanks to all developers and community members for bringing your time and energy!
DeepChem’s Current Design
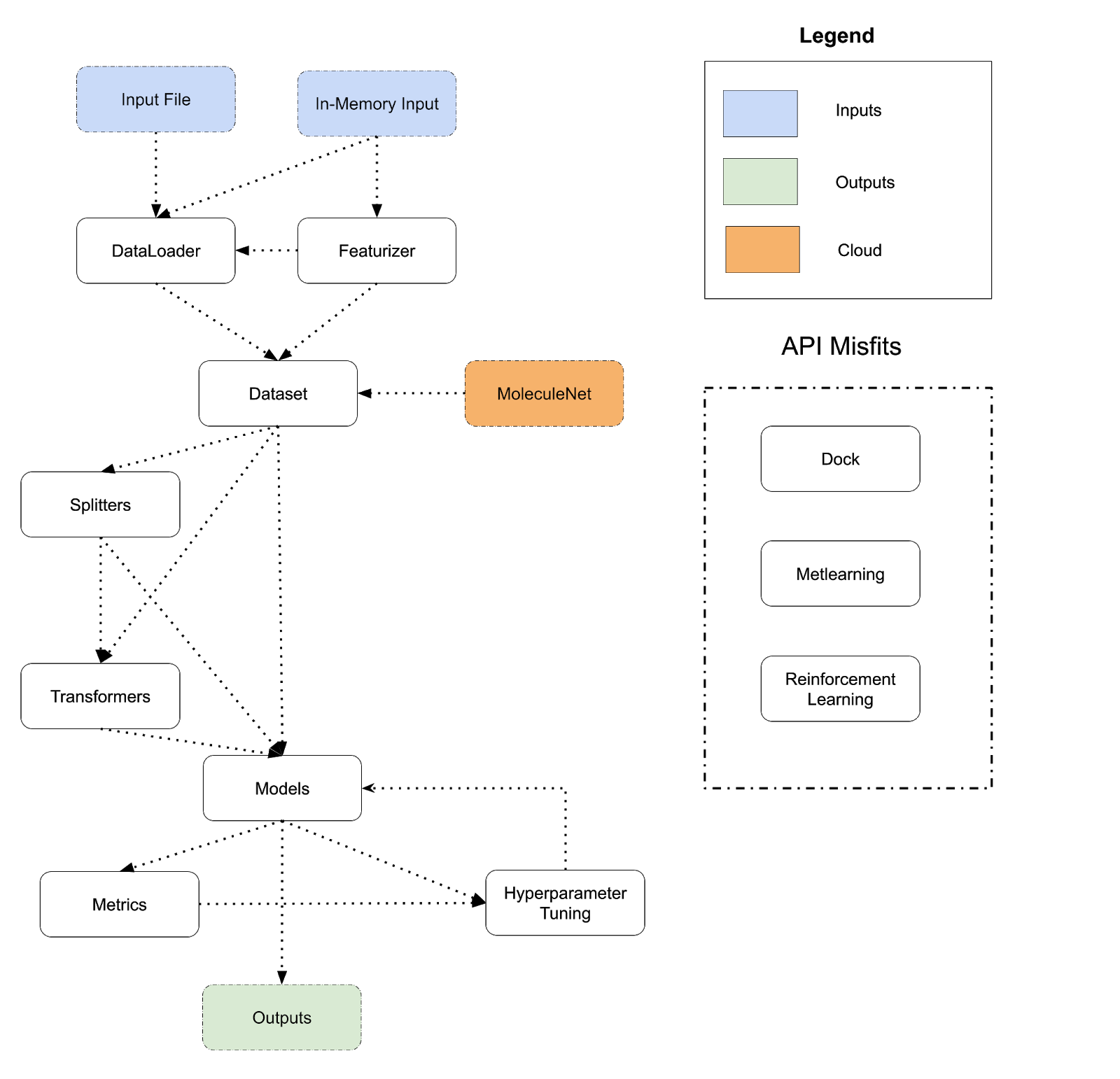
The diagram below lays out the design of DeepChem’s API today. DeepChem programs flow from inputs to outputs and perform a sequence of transformations. There are many different choices to take at each step of this flowchart (for example, there are dozens of different models, many different featurizers, many different transformers and so on), so the flowchart can be expanded into thousands of different systems. The different arrows show different paths that users can take; for example a Dataset can be fed directly into a model or routed through a Splitter and Transformer and then fed into a model. A program corresponds to a path from an input to an output following the arrows.
In effect, DeepChem provides a domain-specific-language (DSL) for scientific machine learning. A sample DeepChem program can be represented in condensed DSL pseudocode as follows (the code below is a conceptual simplification, not actual DeepChem code).
# Conceptual DSL code; Not actual DeepChem code!
input = “filename.csv”
featurizer = Featurizer()
dataset = Loader(input, featurizer)
splitter = Splitter()
train, valid, test = splitter(dataset)
transformer = Transformer()
train, valid, test = transformer(train, valid, test)
model = Model()
metric = Metric()
hyper = HyperparameterTuner()
model = hyper(model, valid, metric)
output = model(test)
Depending on the specific choice of Featurizer/Loader/Splitter/Transformer/-
Model/Metric/HyperparameterTuner we end up with a different program. This basic architecture is very broad and has allowed DeepChem to be applied to a very wide range of different applications (see DeepChem Papers and Discoveries List). At the same time though, there are obvious limitations to this basic DSL structure. To start, DeepChem’s DSL flow is very linear; inputs flow through a series of transformations to produce outputs. We don’t have good support for more complex iterative flows such as pretraining, active learning, or generative models in which the line between model and data is blurred. There are a number of other major limitations with DeepChem’s current design which we outline below
Single Machine Design
DeepChem at present is designed to run on a single machine. We have some limited support for multiprocessing, but no support for working with multiple GPUs or running larger jobs across clusters. The python ecosystem for distributed workloads has improved tremendously with libraries like dask and ray, but DeepChem doesn’t yet leverage these tools to enable large scale featurization, transformation, splitting, hyperparameter optimization and model training.
API Misfits
Referring back to the diagram above, we also see that the dc.dock, dc.metalearning, and dc.rl modules don’t cleanly fit into the existing DeepChem flow and fit awkwardly with the other modules. dc.dock is very special purpose for drug-discovery applications, while dc.metalearning and dc.rl are split into their own submodules, but conceptually are just types of models. Having these dangling API parts doesn’t hurt DeepChem but does make the entire system less conceptually unified.
Lack of Model Composability
DeepChem models are black boxes. There isn’t a convenient method for users to mix and match primitives to construct new models without diving into the internals of model implementation. The situation is worsened by the fact that DeepChem uses a variety of different backend implementations for its models; we currently have models implemented using scikit-learn, xgboost, lightgbm, TensorFlow, PyTorch, and DGL. We will likely add HuggingFace and Jax models as well. This list of backend technologies is unlikely to shrink anytime soon given the sustained investment into new machine learning tools by the broader community. But due to the multiple backends, we have siloed model code that can only be used for one backend, worsening our issues with composability. For example, we have a large collection of Keras model layers which can’t be used with PyTorch/DGL models.
Making DeepChem More Flexible and Scalable
As DeepChem developers, our goal is to broaden the range of useful programs that our users can construct. With each new release, we expand the flexibility and power of the underlying DSL, enabling our users to construct richer scientific programs. Over the last year, we’ve worked to expand DeepChem past its roots as a chemoinformatic library by making splitters, transformers, models and metrics handle non-chemical datatypes. This work is still in progress, but DeepChem 2.5.0 is much more suited to write general purpose scientific machine learning programs than past releases.
The diagram below lays out a potentially expanded design for DeepChem that provides a more powerful framework for scientific programs. This is a preliminary architecture I’m posting to invite review and discussion. I suggest a few major changes to DeepChem’s architecture. First, API misfits like dock/reinforcement learning/metalearning should be integrated more tightly into DeepChem. Docking is the first example of an eventual DeepChem API for simulation. Free energy perturbation could be the next simulation type we support. Constructing a more general framework for running simulations will enable DeepChem programs to implement and use simulations to generate training data. I also propose the addition of a model hub, and subsuming reinforcement learning and metalearning into DeepChem’s models (conceptually, reinforcement learning and metalearning are already models so this unification would fit naturally). Finally, following the lead of popular projects like DGL, I propose creating a DeepChem tensor API with different backends to unify our deep model implementations.
As before, a DeepChem program is a path from input to output following the arrows, but unlike the previous diagram, we’ve added some additional backwards connections. Models can generate datasets (using generative models) and models can power simulations (for example, neural force fields for molecular dynamics).
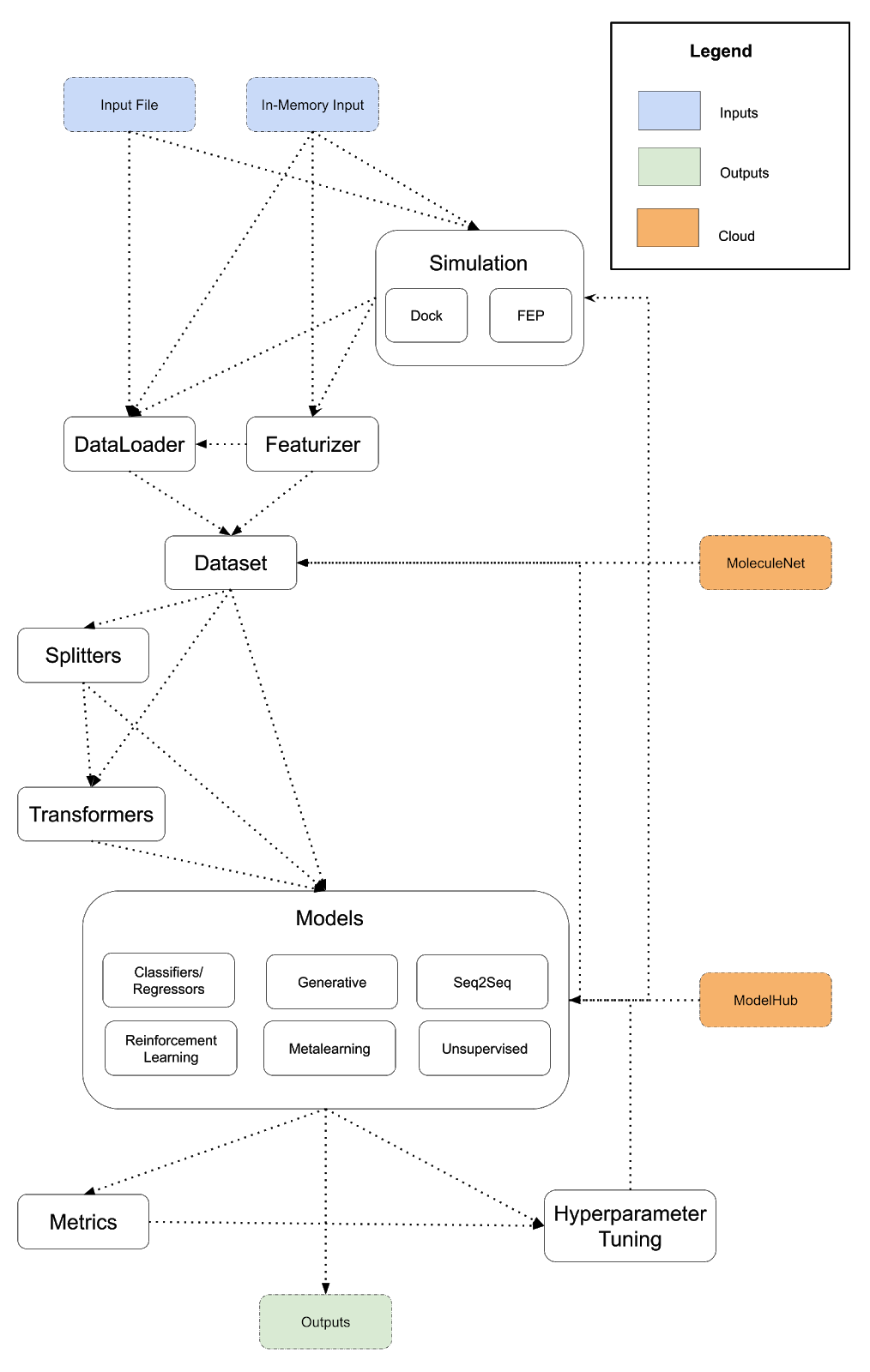
Here is a sample DSL for the expanded vision of DeepChem that runs a simulation to generate a dataset and uses a pretrained model from the ModelHub for training.
# Conceptual DSL code; Not actual DeepChem code!
input = “filename.csv”
# Run simulation to generate data starting from input
sim_results = Simulation(input)
featurizer = Featurizer()
dataset = Loader(sim_results, featurizer)
splitter = Splitter()
train, valid, test = splitter(dataset)
transformer = Transformer()
train, valid, test = transformer(train, valid, test)
# Load pretrained weights from ModelHub
model = Model(ModelHub())
metric = Metric()
hyper = HyperparameterTuner()
model = hyper(model, valid, metric)
output = model(test)
You can imagine making more sophisticated DSL examples that leverage generative models, or use training to improve simulations to create sophisticated feedback loops. We now explain proposed API changes in more depth.
Adding a Simulations Module
Whole ranges of scientific applications depend on simulation. Docking, free energy perturbation, molecular dynamics, fluid simulations, systems biology and many other simulation tools could all be useful to DeepChem users trying to build scientific AI applications. For us to enable these broader use cases, we need a broader framework for using simulation tools with DeepChem. I propose that we add a new dc.sim module that provides tooling for simulations and move dc.dock to dc.sim.dock to become DeepChem’s first simulation API. Over time, DeepChem should grow to support other classes of simulations, starting with free energy perturbation support. At present, we shouldn’t try to mandate a common API for DeepChem simulations, but as our support for new types of simulations grows, we should try to build a sensible common API.
Distributed Computation Support for Featurization/Transformation
DeepChem should more tightly integrate with tools like ray/dask to enable larger jobs. It should become straightforward for a user to kick off a large-scale featurization job (for example) using DeepChem. Underneath the hood, DeepChem should leverage ray/dask or other tools to perform the work necessary for these computations. This functionality will be critical if we want to enable AlphaFold-esque applications in which running featurizations (for protein structure prediction tasks, that means multiple sequence alignment) requires major computational effort.
Multi-GPU and Distributed Training Support
DeepChem models should be trainable with multiple GPUs. New packages and tools like PyTorch Lightning and Keras have made it easy to train models with multiple GPUs. We should aim to have all DeepChem models be able to leverage multi-GPU training. More ambitious would be to support distributed training for very large models. This may require tighter integration with tools like Kubernetes or RaySGD in the longer run.
Standardizing Model APIs
At present, DeepChem’s model infrastructure is mostly geared towards classifiers and regressors. Our core abstract methods in Model are designed for classifiers/regressors, and we don’t have a unified API for other types of models. I propose that we introduce the following abstract subclasses of Model to standardize APIs for other important classes of models:
-
Estimator: The base class for classifiers/regressors -
Generator: The base class for generative models -
Metalearner: The base class for metalearners (moved fromdc.metalearning) -
StructuredOutputLearner: The base class for models with structured output (like Seq2Seq). -
Policy: The base class for reinforcement learning (moved fromdc.rl). -
UnsupervisedLearner: The base class for unsupervised learning methods (such as ChemBERTa pretraining or other language modeling methods)
Standardizing Model Pretraining and Build a Model Hub
Pretrained models are increasingly important for modern machine learning applications. DeepChem should build model hub infrastructure with standard APIs for model pretraining/loading that enables users to easily leverage pretrained models. To start with, I suggest we follow the MoleculeNet model of limiting uploads of pretrained models to DeepChem developers, but it may be possible to open out model uploading to all users in time by partnering Deep Forest Sciences or other companies.
Unifying our Model Infrastructure
At present, we share no code between our models for different backends. We have large amounts of code for Keras that isn’t of any use for constructing PyTorch models. We also lack a clear API for users to compose models/layers. In earlier releases of DeepChem, we supported a custom model building framework called TensorGraph. This framework offered capabilities similar to Keras, but with some additional flexibility. We eventually decided to remove this framework and just support Keras directly since the maintenance overhead was too high. At the time, TensorFlow was our only deep learning framework and Keras had been chosen as the standard API for TensorFlow models, so it seemed like supporting Keras would be good enough for our applications.
In the last couple of years though, the deep learning library situation has continued to evolve. We now support a number of PyTorch models and will likely continue to support both TensorFlow and PyTorch models for several major releases to come. It is also likely that we will support Jax models within the next few releases, and entirely possible that we will support other frameworks as well. The future of DeepChem’s deep learning infrastructure appears to be multi-framework for the foreseeable future.
These changes in the broader ecosystem suggest that it might be useful to revisit our earlier goals with TensorGraph. DeepChem now has a larger community base of developers more able to support custom infrastructure. We also suffer from a fragmentation problem, with an inability to share infrastructure between TensorFlow/PyTorch/Numpy/Jax. Other libraries such as DGL have addressed similar issues by constructing a common tensor API along with different backend implementations for tensor operations (see https://github.com/dmlc/dgl/tree/master/python/dgl/backend). I propose that we consider establishing a common DeepChem tensor API along the lines of DGL’s.
A new DeepChem tensor API wouldn’t change user-facing APIs for DeepChem, but would require us to migrate models to use a common DeepChem tensor standard behind the scenes. This migration can be done gradually over several releases. Once completed, users will be able to use the DeepChem tensor API for their own models. Users should be able to build their own custom DeepChem architectures using the same tools as developers. (Users can of course continue to use KerasModel and TorchModel if they prefer). As developers, we would have to maintain multiple backends, but gain the advantage of a unified model codebase without backend fragmentation. A unified representation may possibly make backend upgrades easier; rather than having to migrate 30 different models to new versions of TensorFlow/PyTorch, we need only migrate the backend tensor implementation. All that said, it’s worth noting up front that some DeepChem models (like DGL wrapped models or scikit-learn/lightgbm models) will likely not be possible to migrate to a shared DeepChem tensor implementation. But enough models should be migratable that we could potentially create a large repository of reusable model infrastructure for our users.
One intriguing possibility is that we can use a common DeepChem tensor API to also power simulations. Jax-md demonstrates how differentiable simulators can be built with Jax. We could potentially support differentiable deepchem simulators in the future. At present, Jax-md is still quite a bit slower than traditional molecular dynamics engines, but as Jax and the underlying XLA compiler speed up, this may start to change over the coming years. Combined with improved distributed computing support through Ray/Dask, we could envision using DeepChem for large-scale differentiable scientific applications.
A DeepChem Maintenance Policy
As DeepChem continues to grow as a library, we want to maintain the fine balance between being a stable platform for production releases and continuing to evolve and grow to address new scientific application areas. I propose that we take the following steps to achieve this goal:
- Breaking Changes Policy: Any API changes in the public-facing DeepChem from 2.5.0 onwards must maintain backwards API compatibility with deprecation warnings for any API changes. A deprecated API must be kept in place for at least a year and can only be removed as part of a DeepChem major version release.
- Long Term Support: As DeepChem continues evolving, we may find large companies that have to use older versions of DeepChem. I propose that the last non-major release before a major version release becomes a long term release. That is, if 2.X is the last version 2 release, 2.X becomes a long term release that will be supported by maintainers for 2 years. This will require us to maintain the 2.X branch on the main repo and perhaps make minor bugfixes if required.
The changes outlined in this document would require major work, likely spanning multiple DeepChem minor and major releases.
Discussion
The ecosystem of scientific machine learning is exploding with hosts of new models and applications. I’d like DeepChem to grow and evolve with its community to support rich ranges of new models and applications. These changes will require DeepChem to shed some of its current rigidity and move towards a more flexible future. In this note, I’ve suggested a number of possible design changes for making DeepChem more flexible.
Part of my inspiration has come from interactions with the Julia community, which has built powerful tools for scientific machine learning (see https://diffeq.sciml.ai/v2.0/, https://github.com/SciML/ModelingToolkit.jl, https://turing.ml/stable/, https://github.com/SciML/DiffEqFlux.jl, https://fluxml.ai/Flux.jl/stable/, https://yaoquantum.org/, https://github.com/NREL-SIIP/PowerSimulations.jl, https://discourse.julialang.org/t/large-scale-hpc-project-on-probabilistic-programming-at-scale-in-conjunction-with-scientific-simulators/39416 and many other efforts). Julia’s scientific developers are often also Julia language maintainers which gives Julia considerable flexibility to evolve its language to better support important scientific applications. In contrast, DeepChem is based on Python and will be for the foreseeable future since that allows us to leverage the vast Python machine learning ecosystem. But, DeepChem can continue to evolve the flexibility and design of its underlying scientific DSL to enable richer classes of DeepChem programs for new applications.
I’d like for DeepChem, working with the broader Python ML community, to enable cutting edge scientific AI research. One day, I’d like for DeepChem to help solve problems in semiconductors, materials, battery design, fluid modeling, systems biology and many other application areas in addition to our core strengths in chemoinformatics and drug discovery. I personally have found DeepChem a powerful tool for my work and I hope we can make it into an even better tool for scientific discovery by making it more scalable, flexible and user-friendly.
Thanks for reading along! Feel free to respond below or on new threads. I anticipate we will need a lot of discussion before we start executing on some of these ideas.